|
Sites de phosphorylation et d'ancrage de phosphatidyl inositol Matériel supplémentaire
|
Breton et al. ont utilisé des approches expérimentales et bioinformatiques afin de déterminer la fonction des protéines COR413. Ils ont ainsi pu analyser leur régulation, identifier les homologues, déterminer la localisation cellulaire, vérifier la structure topologique, trouver des sites de phosphorylation et d'ancrage de phosphatidylinositols glycosylés. Les auteurs présentent également les alignements multiples de la protéine TaVRT-1 ainsi que des résultats expérimentaux, mais ne vont pas plus loin dans leurs analyses bioinformatiques et ne s'aventurent pas dans la prédiction de sa structure 3D. Nous avons donc soumis la séquence de la protéine TaVRT-1 à différents outils de prédiction utilisés pour caractériser cor413 et nous présentons les résultats en les comparant à ceux de COR413-PM et COR413-TM. Nous avons cru bon de compléter ces analyses par une approche de prédiction de la structure tridimensionnelle de la protéine COR413-PM1. PSORT : http://psort.nibb.ac.jp/ Logiciel qui prédit la localisation des protéines dans la cellule. Afin de diriger les protéines aux bons sites les signaux de triage cellulaire obéissent à certaines règles connues(charges, hydrophobicité et patrons) . PSORT applique ces règles à la séquence candidate et détermine la possibilité que la protéine soit localisée à chacun des sites potentiels. Les sites potentiels sont la mitochondrie, le peroxisome, le chloroplaste, le noyau, le réticulum endoplasmique et les régions transmembranaires. iPSORT : http://psort.nibb.ac.jp/ Prédit la localisation cellulaire en détectant les signaux de triage cellulaire N-terminal soient : Signal Peptide (SP), Mitochondrial Targeting Peptide(mTP), ou Chloroplast Transit Peptide (cTP). iPSORT vérifie si la protéine a certaines propriétés particulières , charges des acides aminés et patrons. TargetP : http://www.cbs.dtu.dk/services/TargetP/ Prédit la localisation cellulaire soit secrétion (SP), Mitochondrial Targeting Peptide(mTP), Chloroplast Transit Peptide (cTP) ou autres à l'aide d'un réseau de neurones. SignalP : http://www.cbs.dtu.dk/services/SignalP-2.0/ Prédit la présence de signal peptide et signal d'ancrage à l'aide d'un réseau de neurones et de HMM. Tableau 4. Signaux cibles et localisation cellulaire.
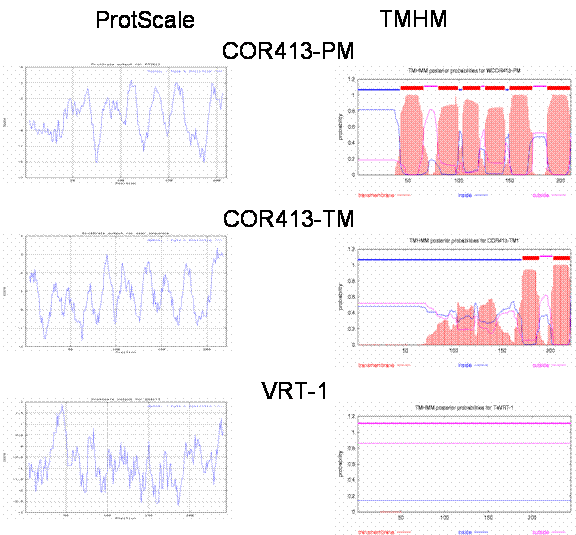
Les prédictions des auteurs sont bien répétées pour COR413-PM et COR413-TM. La seule prédiction pour VRT-1 est celle de PSORT qui prédit une localisation nucléaire ce qui est en accord avec la fonction proposée de facteur de transcription. Protscale : http://www.expasy.org/cgi-bin/protscale.pl Permet de représenter le profil d'une protéine selon différentes propriétés physiques et chimiques des acides aminés telles: hydrophobicité, poids moléculaire, polarité, encombrement et plusieurs paramètres structuraux . TMHMM : http://www.cbs.dtu.dk/services/TMHMM/ Prédit la présence d'hélices transmembranaires dans une protéine. Comme son nom l'indique l'outil fait appel aux modèles de Markov cachés.
Figure 7. Hydropathie et prédictions des domaines transmembranaires. Les résultats pour COR413-PM et COR413-TM correspondent à ceux des auteurs. Les prédictions indiquent clairement la présence de cinq domaines transmembranaires pour COR413-PM. Par contre pour VRT-1 il ne semble pas y avoir de domaines transmembranaires. ScanPROSITE : http://www.expasy.org/tools/scanprosite/ Cherche un patron ou profil reconnu dans la base de données de Prosite. Tableau 5. Résultats de ScanProsite
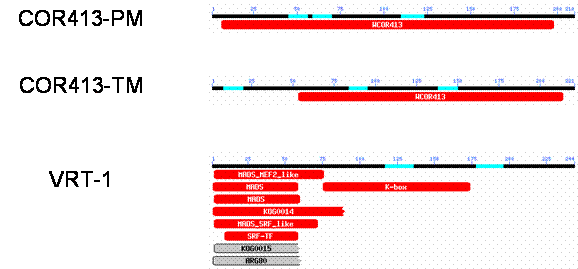
( * [Warning: rule with a high probability of occurrence]) Les auteurs n'ont pas considéré les patrons avec haute probabilité d'occurrence, donc pour COR413-PM et COR413-TM on ne trouve rien d'intéressant . Pour VRT-1 on trouve un patron et un profil correspondant au domaine MADS-box, ce qui n'est pas surprenant. RPS-BLAST : http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi Identifie dans notre séquence les domaines conservés de deux collections soient Smart et Pfam.
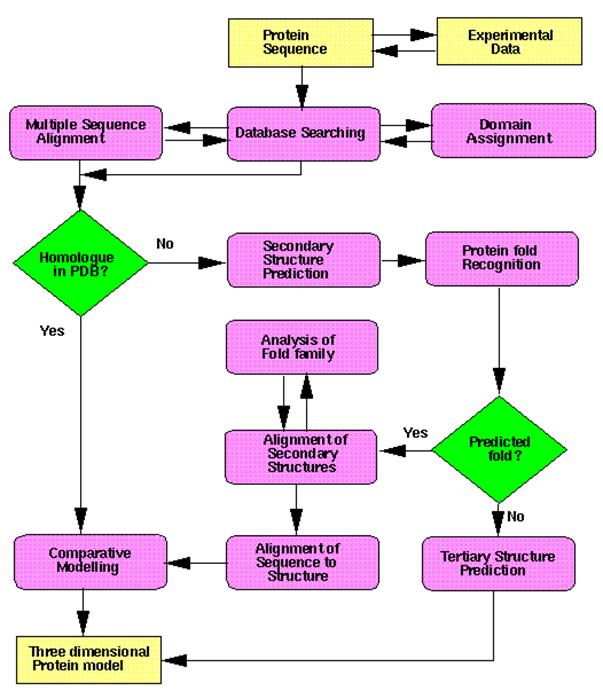
Figure 8. Alignements produits par RPS-Blast. Comme pour ScanProsite aucun résultats probants pour COR413-PM et TM, mais reconnaissance du domaine MADS-box. Prédictions de structures de protéines par analyse comparative La structure 3D des protéines, les coordonnées 3D de tous leurs atomes, est importante à une meilleure connaissance de leur fonction. Elle permet également le design de composés pouvant être utilisés pour leur inhibition en pharmacologie. Les méthodes expérimentales utilisées dans la détermination des structures 3D sont techniquement ardues et couteuses. De plus, de nombreuses protéines ne cristallisent pas facilement (diffraction de rayons-X) ou ne sont pas obtenues en quantités assez importantes (structure par RMN). En date du 6 avril 2004, la base de données de structures des protéines PDB ne contenaient que 25004 structures. Étant donné le nombre de gènes connus, la totalité des structures ne représente qu'un infime partie des connaissances potentielles. Nous avons donc soumis la séquence de la protéine COR413-PM1 à deux outils de prédiction, soient Swiss-Model ( http://www.expasy.org/swissmod/SWISS-MODEL.html ) et 3D-JIGSAW ( http://www.bmm.icnet.uk/servers/3djigsaw/ ). Ces outils basent leurs prédictions sur des similarités des séquences soumises avec des protéines de structures connues. Malheureusement, les deux outils n'ont pu prédire de structure à cause d'une absence d'identité avec d'autres protéines. Nous avons donc utilisé les résultats d'une autre publication du groupe de Fathey Sarhan (Daniluk et al., 2003) afin de procéder à des prédictions de structures 3D. Cette publication caractérise une protéine de vernalisation du blé, TaVRT-1. Les auteurs utilisent des outils semblables à ceux présentés dans le plus haut dans le travail afin d'arriver à leurs résultats. Ceux-ci prédisent que la protéine est homologues aux membres de la famille de facteurs de transcription MADS-box et que son expression est négativement corrélée à celle des protéines COR. La prédiction de structures de protéines par analyse comparative génère des modèles 3D de protéines à partir de la séquence de leurs acides aminés. Le succès de la prédiction demande la connaissance d'au moins une protéine similaire de structure connue. Toutes les méthodes de prédiction par similarité suivent quatre étapes :
Ces étapes peuvent être répétées itérativement jusqu'à l'obtention d'un modèle satisfaisant. Swiss-Model ( http://swissmodel.expasy.org ) est un serveur automatisé de prédiction de structures de protéines par analyse comparative. Il a été mis sur pied en 1993 et est le serveur de prédiction le plus utilisé sur le web. Les structures sont prédites en suivant les étapes décrites plus haut. Afin de sélectionner un patron pertinent, le serveur travaille à partir d'une librairie de patrons de la base de données PDB. Une recherche est menée afin de déterminer si le patron couvre plus d'une région de la séquence soumise. Si c'est le cas, le processus de prédiction est séparé en unités indépendantes. Jusqu'à cinq patrons sont superposés par un algorithme des moindres carrés itératif. L'alignement strutural après filtrage des patrons incompatibles. Un alignement local entre la séquence cible et les patrons est calculée, suivie par une heuristique qui améliore les alignements pour optimiser le modèle structural. Un score est ensuite assignés aux patrons selon leur similarité avec la cible. Comme il est impossible de prédire la structure s'il y a des insertions et des délétions, ces régions sont reconstruites par programmation en espace contraint (constraint space programming, CSP). Le modèle d'abord reconstruit à partir des résidus conservés, puis complété par une étape de minimisation d'énergie. Le serveur Swiss-Model évalue ensuite les résultats par un C-score, qui donne une estimation de la variabilité des patrons à une position donnée. Une description plus en profondeur du mode de fonctionnement est disponible en lisant Schwede et al. 2003. 3D-JIGSAW (http://www.bmm.icnet.uk/servers/3djigsaw/ ) est également un serveur automatisé de prédiction de structures de protéines par analyse comparative. Son mode de fonctionnement est similaire à Swiss-Model (figure 9). Il comporte un mode automatique et un mode interactif, afin d'utiliser les informations structurales sommaires que l'usager peut posséder sur la séquence soumise.
Figure 9. Organigramme du fonctionnement de 3D-JIGSAW. (Tiré de http://speedy.embl-heidelberg.de/gtsp/flowchart2.html ). A
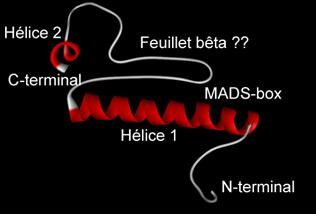
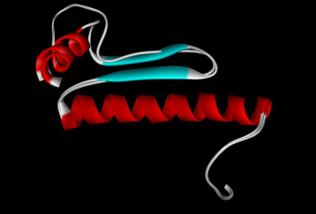
B
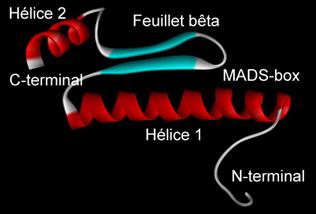
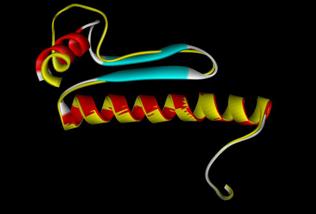
Figure 10. Structure de la protéine TaVRT-1 telle que prédite par A) Swiss-Model et B) 3D-JIGSAW. Les prédictions mettent en évidences deux hélices alpha (Hélices 1 et 2, figure 10), ainsi qu'un feuillet bêta. Le domaine MADS-box se retrouve au début de l'hélice 1. Par contre, il semble que le feuillet bêta soit absent de la prédiction de 3D-JIGSAW. L'allure générale de la protéine est toutefois conservée dans cette région. Alignement des structures 3D prédites Afin d'évaluer les similarités et les différences des structures prédites par Swiss-Model et 3D-JIGSAW, nous avons utilisé l'outil en ligne CE ( http://cl.sdsc.edu/ce.html ). Il utilise un algorithme d'extension combinatoire d'un chemin d'alignement défini par des fragments de paires alignées (AFP). Les AFPs constituent des paires de fragments, un pour chaque protéines comparées, qui confèrent une similarité structurale. Les AFPs se basent sur la géométie locale plutôt que sur des structures globales. Une combinaison des AFPs, représentant des chemins possibles d'alignements, sont sélectivement étendus par extension combinatoire du chemin maximal ou rejetés pour finalement menés à un unique chemin optimal. Une description plus détaillée est disponible dans l'article de Shindyalov et Bourne, 1998. Les résultats de cet alignement sont présentés à la figure 11. A)
B)
Figure 11. Alignements structuraux entre les prédictions de Swiss-Model et 3D-JIGSAW calculés avec l'outil en ligne CE. A) Alignement brut. B) Alignement en colorant la structure prédite par 3D-JIGSAW en jaune afin de faire ressortir les différences. Il est intéressant de remarquer la similarité globale des prédictions des deux outils. En effet, ils prédisent tous deux une longue structure en hélice alpha et une plus courte à l'autre extémité de la protéine (figure 11, structures en rouge). Par contre, les feuillets bêta prédits par Swiss-Model ne sont pas retrouvés par 3D-JIGSAW (figure 11, structures en bleu). Cependant, la structure prédite par 3D-JIGSAW dans la région des feuillet bêta correspond à celle de Swiss-Model. Il existe également des différences dans la plus courte hélice alpha, celle prédite par Swiss-Model ayant deux tours, tandis que celle de 3D-JIGSAW n'en contient qu'un seul. N'étant pas des spécialistes de la structure tridimensionnelle des protéines nous ne nous aventurerons pas dans la signification biologique de ces prédictions.
|
|||||||||||||||||||||||||||||||||||||||||||||