
Séminaire interdisciplinaire de bioinformatique
BIF7002
Projet de session
Modélisation moléculaire 3D et
criblage virtuel
Préparé par :
Azzeddine Saadi
et
Anthony Cowe
21 février 2007
Table
des matières
5. Modélisation Moléculaire 3D
6. Identification de molécules médicamentables
6.1 Filtrage de molécules…………………………………………………………….
6.2 Fonction de score………………………………………………………………...
1.
Introduction
Le processus de découverte de nouveaux
médicaments représente un grand défi pour l’industrie pharmaceutique. Il se
résume essentiellement à identifier de nouveaux composés (molécules naturelles
ou synthétiques) qui vont idéalement évoluer en des médicaments agissant sur
des cibles biologiques spécifiques responsable de disfonctionnements.
L’identification des cibles thérapeutiques est liée à la connaissance du
fonctionnement moléculaire, des voies métaboliques, des systèmes biologiques en
général et à la cause des maladies.
L’achèvement du séquençage du génome humain a
ouvert de grandes perspectives pour l’identification de nouvelle cible
thérapeutique et le développement de nouveau médicaments. Avant l’ère de la
génomique, tous les médicaments commercialisés ont été développé pour environs
500 cibles biologiques connues. Les scientifiques estiment à 3000 le nombre de
nouvelles cibles thérapeutiques qui découleront du séquençage du génome humain.
Dans cette présentation nous allons explorer
une des méthodes utilisée dans le processus de découverte de médicaments à
savoir la modélisation moléculaire et le criblage virtuel. Ces méthodes
d’application in silico touchent plusieurs domaines tels : la
biologie, la chimie, la découverte d’agent thérapeutique, les polymères et même
la nanotechnologie.
2.
Criblage à haut débit
Le criblage à haut débit, en anglais High-Throughput Screening ou HTS
est une méthode d’expérimentation scientifique, il représente une étape très
importante dans le processus de découverte de médicaments et relève des
domaines de la biologie et de la chimie. Il permet de tester un très grand
nombre de composés chimiques (molécules naturelles ou synthétiques) avec des
cibles pharmacologiques afin d’identifier des hits et cela en un temps très
court.
Le criblage à haut débit met en œuvre des
robots capables d’accélérer et d’automatiser des étapes de mise en contact de
molécules potentiellement actives en pharmacologie (candidats médicaments) avec
un système biologique qui reproduit certains aspects de la vie d’une organisme
vivant ou de son dérèglement (maladie). Ce criblage est dit à haut débit car le
processus est très rapide. La technologie du criblage à haut débit est issue des progrès de la biologie
moléculaire, de l’informatique, de la robotique et de la miniaturisation.
Depuis le début des années 90, il est devenu une technique clef de l’industrie
pharmaceutique. Actuellement il existe peu d’outil statistique pour garantire
la détection de hits avec un haut degré de confiance mais beaucoup de
recherches sont entrain de faire afin d’introduire des méthodes statistiques
robustes pour valider les hits et minimiser les faux positifs.
3.
Criblage virtuel
Le criblage virtuel est une technique informatique utilisée en recherche dans le domaine de la conception des médicaments. Il consiste en un parcours test sur de larges librairies de molécules chimiques pour en sélectionner ceux qui ont plus de chance de se transformer en médicaments. Le criblage virtuel est le fruit des avancés scientifiques dans les domaines de la modélisation moléculaire, la chimie combinatoire et la biologie moléculaire. De nos jours typiquement un million de molécules doivent être testé en un courte période de temps, d’où la nécessité d’avoir des méthodes in silico pour faire un criblage rapide et efficace.
Pourquoi faire du criblage virtuel?
Premièrement pour des raisons sientifiques reliées à l’aaugmentation croissante du nombre de cibles, et notamment les cibles 3D, l’augmentation de la puissance de calcul des ordinateurs et une meilleure prédiction des interactions protéine-ligand.
Deuxièmement pour des raisons économiques reliées au coût très élevé du criblage à haut débit ou expérimentale qui n’est pas souvent possible dans le milieu de la recherche et dans les petites compagnies.
Le criblage virtuel se résume en une recherche dans une chimiothèque tridimensionnelle (3D) de molécules satisfaisant aux contraintes d’un pharmacophore et de la structure 3D d’une cible (protéine, ADN, ARN).
4.
Sars
Le sars fait
parti de la famille des coronavirus qui sont des virus d'ARN à brin positif. Ce
virus est caractérisé par de forte fièvre, des malaises, des maux de têtes et
de la toux. Il se transmet par le contact direct entre deux personnes (Anand et
al, 2003). Le 31 décembre 2003, un total de 8096 cas ont été diagnostiqués dans
29 pays et le virus a fait 774 morts. Le Canada n'a pas été épargné avec 251
cas et 43 morts.
Tous les coronavirus codent pour des protéases similaires aux
chymotrypsine qui sont nommées 3CLpro. Ces enzymes capables de faire
la protéolyse sont nécessaires à la maturation des virus (Matthews et al,
1999). Chez le sars la protéase principale s'appelle Mpro et elle
resssemble à celle du picornavirus. Elle clive les polyprotéines pp1a et pp1ab
à aux moins 11 sites qui ont comme séquence leucine-glutamine (L-Q) |site de clivage| suivit d'une sérine
(S), alanine (A) ou glycine (G).

Étant donnée le rôle important de la Mpro dans le cyle
du virus, cette enzyme est considéré comme une cible thérapeutique
intéressante. En effet, en trouvant une molécule homologue à celle-ci, il
serait possible d’inhiber la réplication du virus et ainsi réduire sa
virulence.
5.
Modélisation Moléculaire 3D
La modélisation
moléculaire 3D permet de visualiser et de modifier les molécules dans un
environnement virtuel. Plusieurs informations peuvent être observées sur ces
molécules, telles que : les domaines de liaisons, les hélices alpha, les
feuillets beta et tous les atomes qui les composent. Il est aussi possible
d’obtenir des paramètres comme la charge de la molécule, son énergie minimale
ainsi que les angles et les distances entre les atomes. De plus, les programmes
les plus développés peuvent faire des calculs de mécanique classique, semi
empirique et de mécanique quantique. Toutes ces données permettent de bien
comprendre la molécule étudiée et ainsi, il est plus facile de trouver des
molécules qui lui sont homologues.
Lorsque la structure 3D de la molécule est inconnue, il est possible de
se basé sur d’autre molécule similaire. Dans le cas du sars, il existe des
protéines de la même famille chez les autres coronavirus. Il suffit de
modéliser la Mpro selon les observations faites sur ces enzymes 3CLpro.
C’est à ce niveau que l’information sur struture des molécules est utile (Anand
et al, 2003).
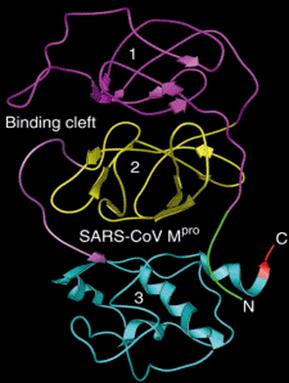
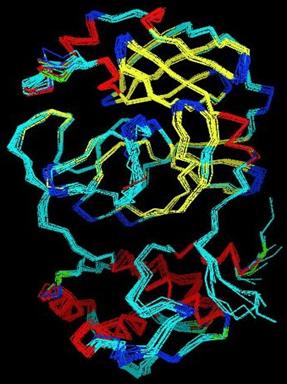
à
droite, il y a les domaines, les hélices alpha et les feuillets beta, tandis
qu’a gauche il y a une superposition de plusieurs modèle de molécules
Une fois la modélisation terminé, il est possible de confirmer la
structure 3D en superposant plusieurs molécules. Par la suite, il est possible
de trouver la molécule inhibitrice qui va se lier au domaine actif ou au
domaine de liaison de cette protéine. Par modélisation 3D, il est possible
d’observer les liaisons et leurs forces pour trouver le meilleur inhibiteur.
Il existe d’autre méthode pour trouver des inhibiteurs, comme la
superposition des ligands naturels. Dans ce cas ci, deux ligands le 1UK4-SARS (vert) et le 1P9U-TGEV (rouge) ont
été obtenus à partir de coordonnée cristalline (voir figure page 6).
À ce ligand est superposé d’autre ligands (6 au total) et cette molécule
est ensuite lié virtuellement à l’enzyme pour vérifier la force des liaisons et
le comporte des deux molécules ensemble.
Finalement, il est possible d’utilisé des inhibiteurs connus pour les
protéines de même famille, puis de les modifier pour qu’ils soient efficace
contre la Mpro. Il y a comme exemple, la molécule KZ7088 qui a été
dérivé de la molécule AG7088 un inhibiteur de la protéase du rhinovirus (Sirois
et al, 2004a).
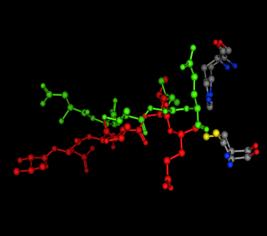
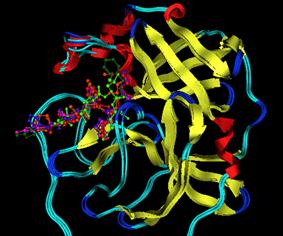
à droite c’est une
visualisation du ligand former de 2 peptides et à gauche c’est le ligand formé
de 6 peptide lié à la protéase.
6.
Identification de molécules médicamentables
Au cours du processus de découverte de médicaments, plusieurs librairies de petites molécules sont criblées afin de déterminer lesquelles ont le potentiel de donner des médicaments. Ces libraires de composés chimiques utilisés dans le criblage à haut débit (HTS) ou le criblage virtuel sont obtenues par plusieurs façons dont des composés obtenus par des efforts précédents d’optimisation de Lead ou dans d’autre cas simplement acheté à des compagnies spécialisées.
La majorité des molécules ne se qualifient pas jusqu'à fin du processus de découverte de médicaments. En fait plusieurs sont éjectés dans l'une ou l'autre des phases de ce processus. Ces échecs font augmenter les délais en moyenne de 10 années et des coûts associés sont de 500 à 800 millions de dollars US. Cette réalité a nuit énormément à l'industrie pharmaceutique et a diminué sa productivité jusqu'à 0.5 nouveaux médicaments par année.
Avec l’achèvement du séquençage du génome humain plusieurs nouvelles cibles thérapeutiques ont vu le jour. Par exemple il est estimé que le génome humain comporte 1000 récepteurs couplés à des protéines G pour approximativement 860 kinases. (Les protéines G de l'anglais guanine nucleotide binding proteins sont des molécules qui permettent le transfert d'informations à l'intérieur de la cellule.) Pour ces cibles potentielles seulement 10% des protéines ont été rationnellement caractérisées (Sirois et al, 2004b)
Afin de pallier à ces problèmes liés au grand taux d’échec dans les différentes phases du processus de découverte de médicaments, plusieurs stratégies sont utilisées pour réduire ce taux échec dont des stratégies de chimie combinatoire ou de la bioinformatique. Plusieurs techniques informatiques et algorithmes ont été introduit, tels que:
l la modélisation pharmacophore
l la modélisation 3D des protéines
l Docking Modeling
l la prédiction de site de restriction sur les séquences des protéines
l la prédiction des classes de familles d’enzymes
l et la prédiction des sites actifs.
De ce fait on s'est rendu compte de l'importance des propriétés physiques des molécules et de l'importance de ne sélectionner que ceux qui sont susceptibles de devenir des médicaments, d'où l'importance pour les compagnies pharmaceutiques et laboratoires de scruter à la loupe les différentes librairies de composés pour leurs propriétés physiques avant de passer au criblage à haut débit ou au criblage virtuel, et toutes les molécules peu probables de devenir des médicaments sont ainsi écartées du processus.
6.1 Filtrage de molécules
Afin d’implémenter ce processus de sélection de molécules, il faut d’abord savoir que-ce qui est un bon Lead et distinguer une molécule médicamentable d’une qui ne l’est pas. Plusieurs critères ont été établis afin d’éviter de faire des testes coûteux et inutiles. Ainsi il y a une convergence de règles qu’un Lead ou un médicament devrait satisfaire afin de les distinguer d’autres classes de molécules, de plus il y a un consensus que les molécules toxiques et réactives et ceux qui ne satisfont pas certains critères physico-chimiques et structuraux soient considérés comme non-médicamentables.
Une des méthodes utilisées pour filtrer les composés qui ont plus de chances de se transformer en médicaments de ceux qui n’en ont aucune, est l’utilisation des fonctions de scores sur les bases de données de molécules.
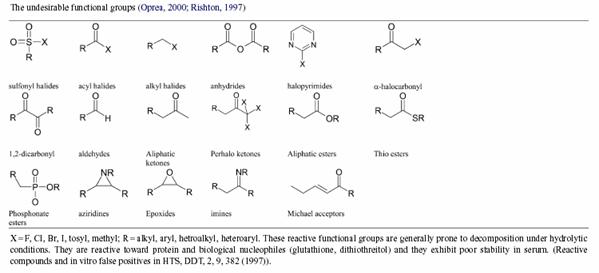
Sirois et Al 2004b, proposent, l’utilisation d’une fonction de score qui prend en compte les propriétés physico-chimiques et structurales des molécules disponibles dans les bases de données aussi bien que la présence de certains groupes fonctionnels indésirables. Cette méthode a été appliquée sur une liste exhaustive de base de données commerciales.
6.2
Fonction de score
La fonction de scores utilisée par Sirois et Al est exprimé en 12 métriques. Deux valeurs sont définies par l’utilisateur, un seuil minimum (cut-off) et un seul maximum. Cette méthode permet de rajouter ou d’exclure des molécules en ajustant manuellement les seuils.
Soit la fonction de
score:

yi représente le descripteur moléculaire à analyser, cut-off
et maximum représentent les seuils minimum et maximum définis pour chaque
descripteur comme dans le tableau suivant :
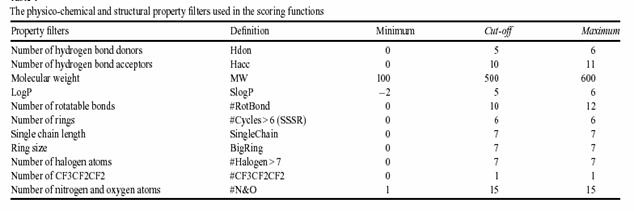
Ainsi la fonction de
score pour les 12 métriques s’écrit :
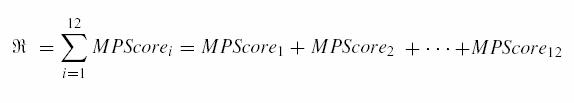
Les facteurs
individuels représentent :
-
MPScore1 = reactive
-
MPScore2 =MW> 500
-
MPScore3 = Hacc > 10
-
MPScore4 = Hdon > 5
-
MPScore5 = logP > 5
-
MPScore6=MW<100
-
MPScore7 = Halogene > 7
-
MPScore8 = CF3CF2CF2 > 1
-
MPScore9 = BigRing > 7
-
MPScore10 = RotBond > 10
-
MPScore11 = (SSSR) cycles > 6
-
MPScore12 = SingleChain > 7
La fonction de score
normalisée pour une molécule s’écrit :
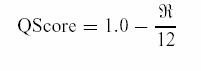
Le tableau suivant désigne les groupes fonctionnels non désirables dans les molécules à tester.
L’échelle de chaque MPScore dépend de la base de données de composés en question afin de permettre aux utilisateurs de faire des expériences en ajustant les valeurs de cuf-off et maximum suivant leurs besoins. Par exemple une base de données peut avoir 200 composés parmi 1000 qui ont un poids moléculaire supérieur à 500 Da, et 150 de des 200 composés qui ont un poids moléculaire inférieur à 525 Da. Une autre base de données peut avoir 200 molécules qui ont un poids moléculaire supérieur à 500 Da mais aussi supérieur à 600. Si on donne la même mesure au descripteur Poids Moléculaire les deux bases de données paraîtront comme si elles avaient le même score alors qu’elles ne le sont pas.
On mesure le score d’une base de données de molécules par l’équation suivante :
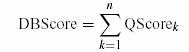
où Qscorek
représente le score normalisé du kième composé dans la base de
données.
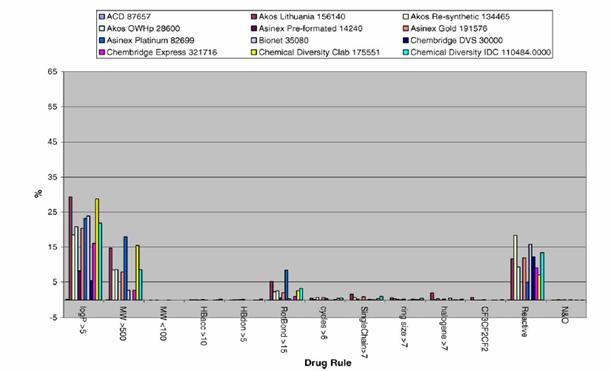
Le tableau suivant
représente un exemple d’un ensemble de base de données aux quelles on a appliqué
la fonction de score de 12 métriques.
7.
Conclusion
Finalement, le criblage virtuel est un domaine
en plein expansion qui va permettre de sauver du temps et de l’argent lors de
la conception de médicaments. De plus, la construction de chimiothèque et de
cibliothèque va faciliter le développement des thérapies futures. Ainsi, il
sera possible de répondre plus rapidement à d’éventuelle épidémie puisque les
molécules thérapeutiques seront déjà caractérisées.
Avec l’informatique, il est aussi possible de
perfectionner les médicaments déjà existant à l’aide des programmes de
modélisation moléculaire. Il suffit parfois de modifier quelques atomes pour
diminuer les effets secondaires d’une thérapie. Ces mêmes programmes peuvent
être utilisés pour adapter un médicament déjà existant à une autre maladie
similaire.
Le cas du sars démontrent bien l’utilité
d’appliquer le criblage virtuel et la modélisation pour accélérer la conception
de thérapie et ainsi obtenir les médicaments plus rapidement.
8. Référence
¾ Anand, K., Ziebuhr, J., Wadhwani, P., Mester, J. et Hilgenfeld, R.,
2003, Coronavirus main proteinase (3CLpro) structure: basis for
design of anti-sars drugs, Science 300
¾ Matthews, D.A., Dragovich, P.S., Webber, S.E.,
Fuhrman, S.A., Patick, A.K., Zalman, L.S., Hendrickson, T.F., Love, R.A.,
Prins, T.J., Marakovits, J.T., Zhou, R., Tikhe, J., Ford, C.E., Meador, J.W.,
Ferre, R.A., Brown, E.L., Binford, S.L., Brothers, M.A., DeLisle, D.M. et
Worland, S.T., 1999, structure-assisted design of mechanism-based
irreversible inhibitors of human rhinovirus 3C protease with potent antiviral
activity against rhinovirus serotypes, proteolytic processing and
physiological regulation, vol. 96, pp. 11000-11007
¾ Sirois, S., Wei, D.-Q., Du, Q., Chou, K.-C., 2004a,
virtual screening for SARS-CoV protease based n KZ7088 phamacophore points,
J. chem. inf. comput. sci., 44, pp.1111-1122
¾ Sirois,
S., Hatzakis, G., Wei, D., Du, Q. et Chou, K.-C., 2004b, Assessment of
chemical librairies for their druggability, computational biology and
chemistry, 29, pp 55-67