Séminaire interdisciplinaire de bioinformatique
BIF7002
Rapport de la conférence du 21 mars 2007
Par Mr. Pierre-Étienne Jacques
Exploitation des données de localisation génomique
(les puces à ADN)
Préparé par :
Yaovi Kounasso
Zahra Foundou
Table des matières
1- Introduction
2- Les puce à ADN
3- Fabrication
d'une puce à ADN
4- Les domaines d'application des puces à ADN
5- Les Applications des puces à ADN
- 5.1 les facteurs de
transcription
- 5.2 la
localisation des sites d'intéraction de protéines avec la chromatine
- 5.3 les nucléosomes
- 5.4 les protéines histones
- 5.5 modification des histones
- 5.6 localisation des H2A.Z
- 5.7 problèmes liés aux
spots
6- Les analyses
statistiques
7- Conclusion
Bibliographie et références
1. Introduction
À l'instar des méthodes traditionnelles de localisation des gènes (
exemple Southern blot, Nordern blot) qui ont leurs champs d'action limités à
quelques gènes, la technologie de puces à ADN permet de mesurer simultanément
et quantitativement l'expression relative de plusieurs milliers de gènes, voir
du génome.
Dans ce rapport, suite à la présentation de Monsieur Jacques, nous allons dans
un premier temps mettre l'accent sur les technologies et l'utilisation des
puces à ADN. Nous attarderons ensuite sur certaines applications de cette
technologie en nous basant sur des exemples précis. Pour en finir nous serons
conduits à analyser des données obtenues en utilisant des puces à ADN à haute résolution.
2. Les puces à ADN
Décrite en 1995 par Schena M et al. La technologie de puces à ADN
est devenue un outil majeur de recherches, incontestable dans la communauté
scientifique. Cette technologie de pointe qui combine les avancées de la
technologie de l'information et les acquis de la science de la vie, permet en
un temps record de présenter des résultats fiables, précis et intéressants.
2.1. définition d'une puce à ADN
Est un ensemble de molécules d'ADN fixés
sur une surface qui peut être du verre, du silicium ou du plastique. Cette
biotechnologie récente, permet de visualiser les gènes exprimés (transcrits
dans une cellule d'un tissu donné.
|
Lame de verre (microarrays) |
Filtres haute densité (macroarrays) |
Puces à oligonucléotides |
|
|
|
|


fig1: Les différentes sortes de puces ;
extrait de DNA microarray principale
Les méthodes de
puces à ADN reposent sur les techniques d'hybridations simultanées sur un
support de taille réduite (quelques cm2), utilisant de l’ARN comme
substrat.
Bien que partant d’un principe de base commun, certains puces à ADN se
distinguent sur trois (3) points essentiels :
la nature du support le support peut être en verre ou en nylon.
le système de détection : Radioactivité, colorimétrie,
fluorescence.
Le choix des sondes et la technique d’hybridation: L’utilisation
des fragments de ADNc ou des oligonucléotides et le choix du système de détection,
contribuent à connaître les différents microarrays.
Le
support
Le support le plus utilisé est réalisé sur des lames de verre d’un format
allant de 1 à 10 cm2. Le choix de lames de verre se justifie dans la
mesure où ce support permet une approche miniaturisée, avec de très hautes
densités (plusieurs dizaines de milliers de spots par cm2) et est
adapté pour de faibles quantités d’ARN.
Les filtres en nylon sont moins coûteux, mais moins utilisés. Malgré leur
pouvoir d’être réutilisés et leur grande sensibilité, la densité des spots est
limitée.
Le système de détection
La technique de détection la plus répandue est la fluorescence. Elle permet une
bonne sensibilité de détection du signal tout en étant facile à manipuler
contrairement à la radioactivité.
Notons que deux types d’approches sont envisagés en fonction de l’utilisation d’un
ou deux marqueurs.
L’approche non compétitive : utilise deux échantillons qui sont
marqués avec le même fluorochrome, exemple phycoérythrine pour la technologie
Affymetrix. Chacun des échantillons est hybridé sur une puce séparément. L’intensité
du signal est proportionnelle à l’expression du gène correspondant.
L’approche compétitive: Est basée sur la technique d’hybridation
compétitive.
Les deux échantillons sont marqués chacun avec un fluorochrome différent, par
exemple cyanine 3-CTP (Cy3) et cyanine 5-CTP (Cy5). C’est la stratégie adoptée
par Agilent Technologies et autres fournisseurs.
Les deux échantillons sont mélangés en proportions équivalentes. Lors de l’hybridation
sur la puce il y a compétition pour l’appariement à la sonde immobilisée sur le
support solide. De cette manière on mesure directement un ratio entre le signal
fluorescent émis par l’échantillon marqué en Cy5 et celui de l’échantillon
marqué en Cy3.
2.2. le choix de séquences sur les spots et le type
d'hybridation
Les puces à ADN :
Les fragments de ADNc utilisés sont de quelques centaines de bases. Ils sont
immobilisés sur les supports par des liaisons chimiques covalentes. Un désavantage
de cette méthode est que, ces fragments d'ADNc ne correspondent pas nécessairement
à des gènes identifiés. L'utilisation de banques de données nécessite un
stockage soigneux des clones de ADNc et des produits d'amplification utilisés.
De plus il faut un contrôle minutieux des séquences, ce qui est parfois
fastidieux.
Les puces à oligonucléotides :
Les sondes sont des oligonucléotides de synthèse de 25 à 80 bases. Les séquences
choisies sont optimales pour hybrider chaque gène-cible (intensité, sensibilité
et spécificité). Notons que ce type de sondes est utilisé uniquement pour la détection
de gènes connus séquencés.
3.
Fabrication d'une puce à ADN
- des fragments d'ADN amplifiés par la
technique PCR, déposés sur une lame de microscope recouverte de polylysine
(sonde)
- la polylysine assure la fixation de l'ADN
- l'ADN est dénaturé avant l'hybridation, pour
qu'il se trouve sous la forme simple brin sur la puce.
- l'accrochage est effectué au brin complémentaire
contenu dans la cible
- NB/ il existe d'autres tubes de puces que
celles sur lames de verre :
- filtres haute densité (macroarrays)
- lames de verre (microarrays)
- puces à aligonucléotides
- les molécules d'ADN fixés sur la lame sont
appelées des sondes
- des milliers de sondes peuvent être fixés
sur une même puce.
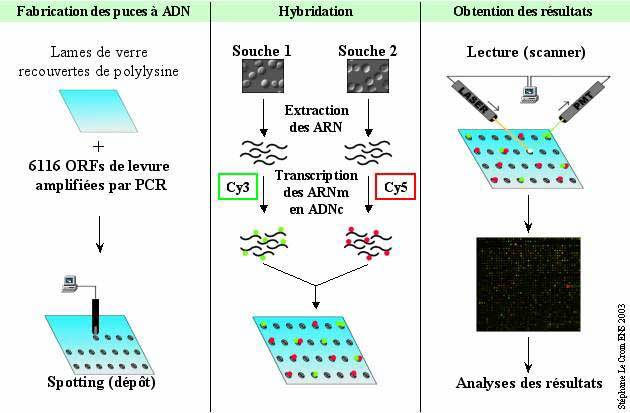
fig2: Les étapes de fabrication de puces à ADN; Extrait du doc. de Stéphane
le Crom et Philippe Marc
3.1.
Hybridation
§
les ADN (vert) et les ADN (rouges) sont mélangés (cibles) et placés
sur la matrice simple brin déposés (sonde)
- la puce est inculpée une nuit à 60o
§
à cette température, un brin d'ADN qui rencontre son complémentaire
s'apparie pour donner de l'ADN double brin
- les ADN fluorescents vont s'hybrider sur les
gènes déposés
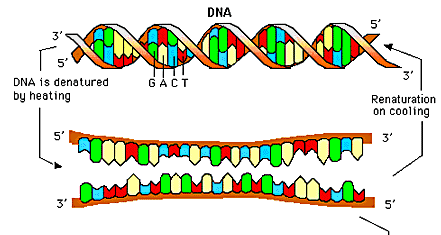
fig3: Hybridation ADN ; Extrait du doc. de Stéphane le Crom et Philippe
Marc
Un lien vers une animation:
http//:www.Cea.fr/UserFiles/File/Animations/aLaLoupe/biopuce/puceAdn.htm
4. Les domaines d'application des puces à ADN
Les domaines
d'applications des puces à ADN sont très vastes et touchent un large marché tel
que :
- la recherche biologique ( génomique
fonctionnelle)
- la recherche pharmaceutique
- le génotypage
- le diagnostic des maladies
- les contrôle agroalimentaires et industriels
- l'environnement
- identification et expertise médico-légale
5. Application des puces à ADN
5.1.
Les facteurs de transcription
Lors de la comparaison d’une expérience des profils d’expression
entre une souche sauvage et une souche mutante pour un facteur de
transcription, on remarque souvent qu’une portion importante des gènes
différentielle ment exprimés est souvent le résultat d’effets indirects.
Tandis que la localisation génomique, appliquée par exemple au même facteur de
transcription permet quant à elle d’identifier les cibles directes.
Impact (indirect ) de la mutation d'un
facteur de transcription dans une expérience de comparaison de profils
d'expression
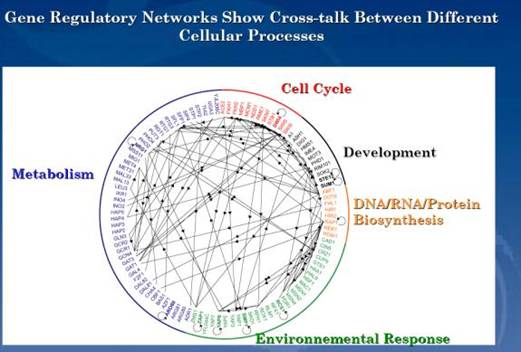
fig4: Régulation
de gènes ; extrait de l'exposé de
J. Étienne Jacques
Tous les facteurs
de transcription (à la périphérie du cercle) ont été localisés directement sur
le promoteur d'autres facteurs de transcription pointés par les flèches (le
promoteur de plusieurs autre gènes est également lié directement mais seulement
les interactions impliquant des facteurs de transcription sont montrées) .
Structures des réseaux régulateurs entre les différents
facteurs de transcription
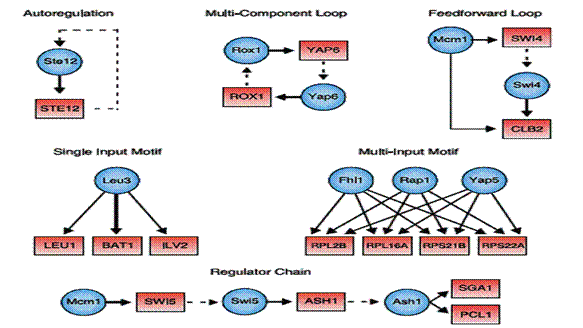
fig5: Structure
des réseaux régulateurs ; extrait
de l'exposé de J. Étienne Jacques
Par exemple, le facteur de
transcription Rox1 (cercle bleu) s'attache directement sur le promoteur du gène
YAP6 (rectangle rouge) pour le réguler, alors que sa protéine Yap6 (aussi
facteur de transcription) régule en boucle de façon directe le gène ROX.
5.2.
La localisation des sites d'interaction de protéines avec la chromatine
La méthode ChIP-on-Chip (Chromatin-ImmunoPrecipitation on Chip), a été
inventée dans le but d’identifier des sites d’interaction de protéines
avec l’ADN génomique.
Cette méthode est plus utilisée chez la levure du boulanger. Pour
des raisons suivantes :
Le génome est de taille restreinte et la faible proportion des régions
intergéniques facilite la construction de puces contenant ces régions, qui sont
les cibles de facteurs de transcription.
Quelques études
faites sur les facteurs de transcription.
Une étude de grande envergure sur plus de 100 facteurs de
transcription marqués d’un épitope Myc (sur 161 protéines décrites dans la base
de données YPD 7 comme ayant une activité de liaison à l’ADN et de
transcription) a réalisé en 2002
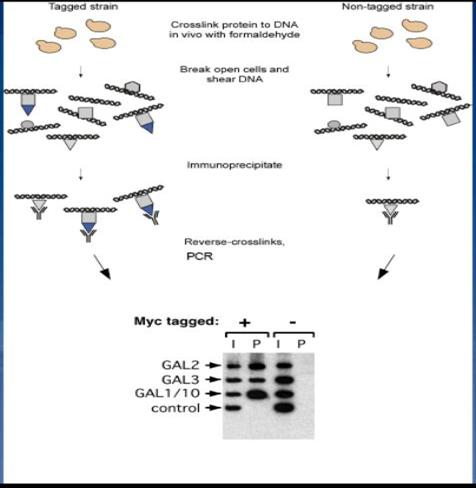
fig6: Interaction de protéines avec ADN; extrait de l'exposé de J. Étienne Jacques
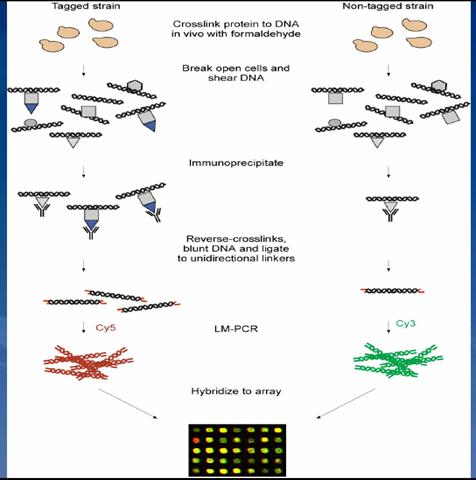
![]()
fig7: Interaction de protéines avec ADN ; extrait de l'exposé de J. Étienne Jacques
Principe du « ChIP-on-Chip
Principe du
fonctionnement de ChIP-on-Chip
Fixation des protéines de manière covalente à l’ADN génomique par un traitement
au formaldéhyde
Fragmentation
de l’ADN et incubation de l’extrait cellulaire avec un anticorps spécifique de
la protéine d’intérêt (ou spécifique d’un
épitope avec laquelle la protéine aurait été marquée au préalable).
La purification par immuno-précipitation permet d’isoler la protéine d’intérêt
avec les fragments d’ADN qui lui sont associés.
5.3. Les Nucleosomes
La substance
chimique qui constitue les chromosomes s’appelle la chromatine. C’est un mélange
de DNA et de protéines. Cette substance est composée de particules appelées « nucléosomes »
empilées les unes au-dessus des autres selon le modèle dit en «
collier de perles».
5.4. Les Protéines Histones (cœur du nucléosome)
Ce sont des protéines basiques, riches en acides aminés tel que l’Arginine
et la Lysine. Elles sont donc riches en charges positives ce qui leur permet de
former des liaisons ioniques avec le DNA (qui porte de charges négatives dues
aux fonctions acides libres des molécules d’acide phosphorique). Le cœur de L’histone
est formé en réalité d’un octamère comprenant 2 copies des histones H2A, H2B,
H3 et H4. Une 5e Histone, H1, ne fait pas partie du nucléosome mais
interagit avec le DNA de 2 nucléosomes contigus. Les histones peuvent subir des
modifications covalentes réversibles : acétylation, phosphorylation,
ribosylation, l’ubiquitination. Cette possibilité de changement rapide et réversible
joue certainement un rôle important dans la physiologie.
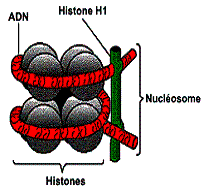
fig8: Organisation des histones; extrait de obiweb.bcgsc.ca/medgen
H1 servent à relier les nucléosomes les uns aux autres de façon à forme une structure spirale plus compact.
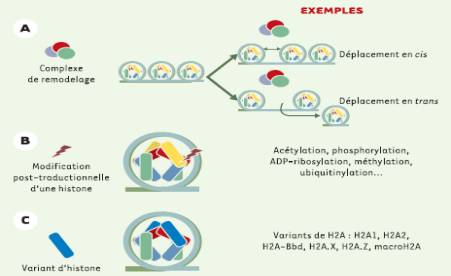
fig9: Les voies
de modification du nucléosome ;
extrait de Md/Sc 203; 19:1137-45
Les trois voies de modification du
nucleosome
a) déplacement en cis ou en trans. Des nucleosomes par un complexe de
remodelage.
b) modification post-traditionnelle des extrémités des histones
c) substitution d une histone conventionnelle par un variant.
5.5. Modification des Histones
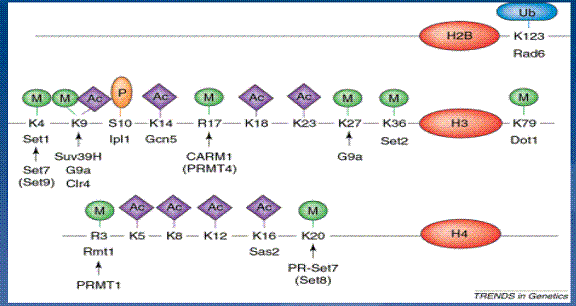
fig10:
Modification des histones; extrait de l'exposé de J. Étienne Jacques
Ce schéma nous renseigne sur différentes modifications dans
les histones : méthylation (M), acétylation (Ac),phosphorylation (P),
ubiquitination (Ub)) caractérisées sur quelques-uns des résidus (lysine (K), sérine
(S), arginine (R)) des queues des histones H2B, H3 et H4. Ces modifications
jouent des rôles biologiques très distincts, qu'il est par conséquent très
important de pouvoir les caractériser indépendamment. Il existe présentement
des anticorps très spécifiques permettant leur localisation génomique.
5.6.
Localisations de H2A.Z (chez la levure de boulanger)
Le gène qui
code pour la H2A.Z est retrouvé chez certains organismes comme S. pombe, S. cerevisiae et Tetrahymena
Sa transcription est contrôlée au cours du cycle cellulaire et dépend en
partie de la réplication.
Notons bien que chez les mammifères, son expression de la H2A.Z, n’soit pas couplée de la synthèse
de l’ADN.
La séquence peptidique du variant
H2A.Z présenterait 60% d’homologie avec
celle de l’histone H2A et 90% d’homologie inter-espèce. Cette variante jouerait
un rôle très important dans des fonctions spécifiques liées à la prolifération,
la différenciation et le développement
Pour en conclure, H2A.Z semble jouer un rôle à la fois positives et
négatives sur l’expression des gènes.
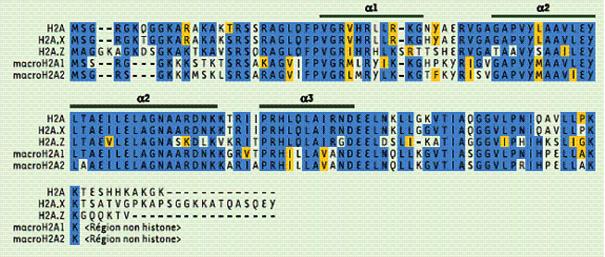
fig11:
Comparaison des séquences des histones humaines; extrait de Médecine/Sciences
203; 19:1137-45
Comparaison
des séquences des histones humaines de la famille H2A
Les résidus identiques sont figurés en bleu et les résidus similaires en jaune.
L’alignement
a été réalisé avec le programme CLUSTALW .
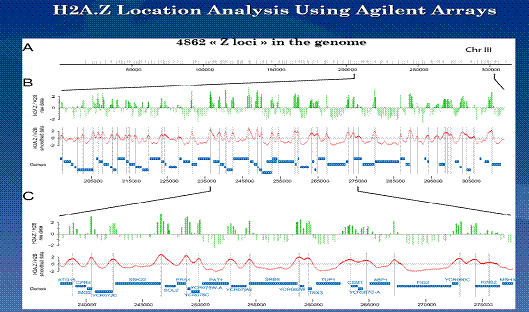
fig12:
Localisation des H2A.Z ; extrait de
l'exposé de J. Étienne Jacques
Chaque barre verte représente
l'emplacement d'un oligonucléotide spécifique dans le génome. L'intensité représente
l'enrichissement de chacun de ces emplacements pour la variante d'histone
H2A.Z. On remarque la présence d'un maximum d'enrichissement en amont de chaque
gène (dans le promoteur) alors qu'on ne retrouve pratiquement pas de H2A.Z dans
les régions codant et en aval des gènes.
5.7 Problèmes liés
aux spots
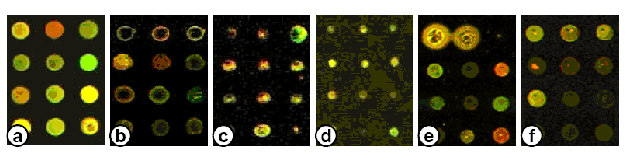
fig13: Défauts de spots; extrait de UMR144 CNRS – Institut Curie
(a) spots réguliers ; (b) spots en anneaux ; (c) spots irréguliers
dus à un mauvais calibrage du robot ;
(d) spots trop petits ; (e) spots gonflés dus à une mauvaise
purification des produits de PCR ;
(f) particules rouges dans les spots indiquant une surface de polylysine
abîmée.
Pour éviter ces problèmes liés aux spots, toujours
dans le souci de localiser tous les
gènes identifiés et d’économiser le temps et l’argent, l’équipe de recherche dans
laquelle travaille M. Jean Étienne
Jacques a dû fabriquer de plus
des spots adaptés aux exigences de leur travail. Ces puces sont non biaisés
mais force est de constater que selon les explications données par M. Jacques, on note certains non-appariements.
6.
Les analyses statistiques
Filtrage primaire des données : Il
existe différents types de logiciels d’extraction permettant de repérer
visuellement les spots non exploitables. Ceci se fait grâce à un système de
balisage qui assigne un code numérique selon la qualité du spot considéré, afin
de faciliter le filtrage des données non significatives.
C'est l'exemple du logiciel GenePix Pro, où le chiffre « 0 »
correspond par défaut à un spot exploitable, « –100 » à un spot
défini mauvais « –50 » non détecté par le logiciel et « –25
» à un spot défini vide par l'utilisateur.
Le calcul des ratios d’expression :
Il existe plusieurs méthodes de calcul du ratio d’expression: ratio des
intensités moyennes, médianes, régression linéaire.
Notons que le niveau d’expression relatif de chaque gène est estimé par la
valeur du ratio d’expression calculé à partir des intensités des signaux en
rouge et en vert.
A = intensité médiane du spot i en rouge C = intensité médiane du
spot i en vert
B = médiane du bruit de fond local en rouge D = médiane du bruit de fond
local en vert
|
|
Rr/v (i) = (A-B)/(C-D)
Pour manipuler et analyser les données, les
ratios d’expression sont usuellement transformés dans une échelle logarithmique
: log2(r/v). La raison de cette transformation est que le logarithme permet de
transformer le ratio d’expression en une fonction linéaire, symétrique de –
l’infini à + l’infini, centrée sur 0 et additive, ce qui facilite les analyses
statistiques.
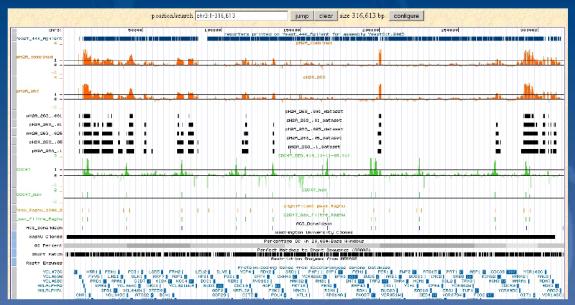
fig14: Analyse statistique ; extrait de l’exposé de J-Étienne Jacques
Sur le
schéma du logiciel d’analyse, on voit certaines espaces vides (ligne bleue en haut). Cela
représente les données non significatives, dues soit aux problèmes de défaut de spots, ou aux
problèmes liés à des contaminations, ou
soit aux problèmes de réglage de lumière (intensité)….
Il est très regrettable que l’on
ne puisse pas tester le fonctionnement de ce logiciel pour mieux comprendre le
bien fondé des choses.
7.
Conclusion
Le défi dans
la réalisation d’un projet de puces à ADN est de pouvoir garantir la fiabilité du
processus, tout en augmentant les performances et en assurant la sécurité
analytique de l’ensemble. Un bon choix des matériels, s’assurer d’une bonne
interprétation des données et des
analyses statistiques afin de
fournir une bonne documentation.
Malheureusement
les réponses attendues ne sont pas toujours à la hauteur, car certains facteurs
tels que l’environnement, les contaminations, les problèmes techniques pour n’en citer que ceux
là, rendent les choses difficiles. Et cela pourrait être un obstacle à
l'utilisation des puces à ADN en terme d’application clinique. Mais l'avenir
est promoteur.
On s'attend
un jour à ce que les puces à ADN deviennent des vrais outils utilisés dans des
recherches cliniques.
L’exposé de M. Jacques, en générale a été intéressant. Nous le remercions pour
les aspects théoriques et pratiques enrichissants, pour l’explication apportée
sur sa propre conception des puces (non biaisés ), la fabrication de ces puces au Labo et l’utilisation de
cette technologie pour ses recherches réussies.
Malheureusement
ne sommes pas en mesure de tester les logiciels utilisés par M. Jacques pour
les analyses statistiques.
Références