Rapport
de conférence de Henry Xing :
Quelques
mesures pertinentes pour calculer la distance entre les communautés d'espèces
dans des réseaux de similarité de séquence
Fait par : Gabrielle Boudreau et Stéphane
Samson
Dans le cadre du cours : BIF7002
INTRODUCTION
La phylogénie est l’étude des relations
d’évolution entre les organismes.1 Ce domaine repose sur l’hypothèse
que tous les êtres vivants descendent d’un ancêtre commun.1 À partir
de cet ancêtre commun, des évènements de spéciation tels l’isolation d’une
population ou l’adaptation à un nouvel environnement peuvent entrainer ses
descendants vers des branches évolutives divergentes.2 La phylogénie
tente de reconstruire l’histoire de ces spéciations successives durant
l’évolution en reconstituant les filiations évolutives à partir de données
génétiques.1 En assumant que le taux de mutation génétique reste
constant tout au long de l’histoire biologique, le nombre de mutations identifiées
entre deux individus, populations ou espèces peut servir d’horloge évolutive et
aider à déterminer quand la séparation s’est produite.1 Depuis
l’origine des espèces de Darwin, les arbres phylogénétiques ont été largement
utilisés comme représentation schématique de l'évolution.1
Arbre phylogénétique
Ce graphe connexe acyclique d’ensemble de
nœuds est connecté par des arêtes de telle sorte que toute paire de nœuds sont
reliée par exactement un chemin (Figure 1).3 Les êtres les plus
proches ne divergent que dans les dernières branches de l’arbre, tandis qu’une
ancestralité commune éloignée est marquée par un positionnement sur des
branches très divergentes. À partir de ces arbres, il est possible de calculer
différentes mesures de distance évolutive, tels que la distance de Robinson et
Foulds, des quartets, ou encore la distance UniFrac. Cependant, une multitude
d’études ont démontré que les arbres phylogénétiques ne permettent pas de bien
représenter le transfert génétique latérales.1,4,5 De plus, une
autre des limites de ces modèles évolutifs simplifiés est leur capacité de
représenter l'hybridation et l'introgression entre les espèces et la
recombinaison de diverses formes.6-8 Ces limites reconnues des
arbres phylogénétique ont motivé l’adoption de représentation graphique
alternative, les réseaux.3,9
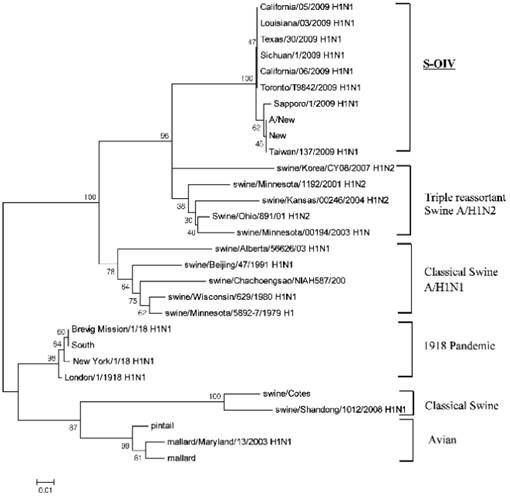
Figure 1 : Représentation d’un arbre phylogénétique. Cet exemple tiré de Mondal et al. représente l’arbre phylogénétique des
différentes souches d’influenza H1N1 et H1N2, basée sur la protéine hémagglutinine
virale (HA).
Réseau phylogénétique
Les réseaux phylogénétiques sont adaptés
pour représenter les phénomènes évolutifs complexes et ont permit l’émergence
de nouveaux calculs de distance évolutive.10-13 Ceux-ci sont une
méthode de modélisation des relations entre diverses composantes représenter
respectivement par des arêtes et des nœuds (Figure2).14 Dans un
contexte phylogénétique, les composantes, soient une séquence, un gène ou un
génome d’un organisme, sont généralement représenté par un nœud tandis que les
arêtes démontrent une similarité entre les séquences supérieure à un seuil
donné. La similarité est une valeur qui se mesure en pourcentage d'identité,
l’identité elle même définie comme le niveau de ressemblance entre deux
séquences.
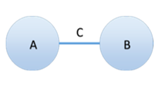
Figure
2 : Schématisation des éléments d'un réseau où A et B représente un nœud et C
représente une arrête.
Distances évolutives
Henry Xing et ses collaborateurs ont
développé cinq nouveau calculs de distance évolutive pouvant être calculés à
partir de réseaux phylogénétiques : NetUniFrac, Spp, Spep, Spelp
et Spinp. La distance NetUniFrac est la
généralisation pour les réseaux de UniFrac, une distance évolutive calculé sur
les arbres phylogénétiques.
UniFrac qualitative est une méthode
phylogénétique mesurant la distance entre les communauté biologique (Formule
1).15 Cette méthode est une fraction de la longueur des branches
dans un arbre phylogénétique qui mène aux descendants d’un échantillon ou
l’autre, mais pas les deux. Elle permet de déterminer si les communautés
biologiques sont significativement différentes, identifier les lignées
individuelles qui contribuent à ces différences et révéler des schémas généraux
reliant de nombreux échantillons.15
![]()
Formule
1 : Formule de distance d’évolution UniFrac
qualitative où Uab est la distance UniFrac
entre l’échantillon A et B, unique est la somme de la longueur des branches qui
mène à des espèces d’une seule communauté et total est la somme de la longueur
de toutes les branches.
NetUniFrac, tant qu’à elle repose sur les
arêtes monochromes (Formule 2). Une arête est dite monochromes si à ses deux
extrémités, les espèces associées appartiennent à la même communauté. Ce calcul
a pour avantage d’être calculée dans un temps linéaire par rapport au nombre
d’arêtes du réseau.
![]()
Formule 2 : Formule de distance d’évolution NetUniFrac
où ssnUab est la distance NetUniFrac,
monochrome est la longueur totale des arêtes monochromes du réseau et total est
la longueur totale des arêtes du réseau.
Les quatre calculs de distance, Spp, Spep, Spelp
et Spinp, sont basé sur le chemin le plus court. Ce
paramètre de centralité des nœuds indique la place de ce nœud par rapport aux
autres nœuds du graphe. Dans un réseau hautement centralisé, il y a convergence
vers une composante centrale qui mène à plusieurs spéciations. Un réseau moins
centralisé, quant à lui ne possède pas de point central. Le chemin le plus
court entre deux nœuds a et b correspond à la séquence de nœuds connectés par
des arrêtes tel que le nombre de nœuds intermédiaires est minimal dont les
extrémités du chemin sont les nœuds a et b. Le chemin le plus court est donc
définies par le diamètre du réseau.16
La distance Spp («Shortest path
proportion») est la proportion des chemins les plus court qui sont monochrome
par rapport à l’ensemble des chemins du réseau. (Formule 3)
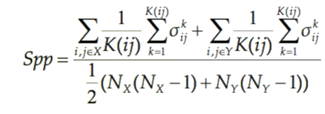
Formule 3 : Formule de distance
d’évolution Spp où Spp est
la distance évolutive Spp, x et y sont deux
communauté d’espèce, K(i,j)
est le nombre de chemins entre les nœuds i et j, NX et NY
sont le nombre de nœuds dans les communautés X et Y. Si le chemin K entre
les nœuds i et j est monochrome ![]() est de 1 sinon 0.
est de 1 sinon 0.
La distance Spep
(«Shortest path edge proportion») est basé
sur le nombre d’arêtes monochromes dans tous les chemins les plus courts entre
les espèces d’une même communauté (Formule 4).
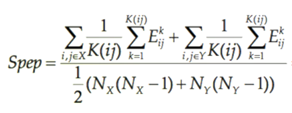
Formule 4: Formule de distance d’évolution
Spep où Spep est la
distance d’évolution Spep, x et y sont deux
communauté d’espèce, K(i,j) est le nombre de chemins
entre les nœuds i et j, NX et NY sont le nombre de nœuds
dans les communautés X et Y. ![]() est la proportion d’arêtes monochromes
dans le chemin k entre ij les nœuds i et j
appartenant à la même communauté.
est la proportion d’arêtes monochromes
dans le chemin k entre ij les nœuds i et j
appartenant à la même communauté.
La distance Spelp
(«Shortest path edge length
proportion») est basé sur la longueurs des arêtes dans tous les chemins les
plus courts monochromes (Formule 5).
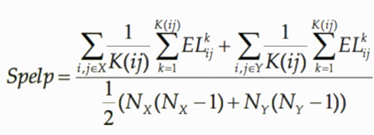
Formule 5: Formule de distance d’évolution Spelp
où Spelp est la distance d’évolution Spelp, x et y sont deux communauté d’espèce, K(i,j) est le nombre de chemins entre les nœuds i et j, NX
et NY sont le nombre de nœuds dans les communautés X et Y. ![]() est la proportion des longueurs d’arêtes monochromes
dans le chemin k entre les nœuds i et j appartenant à la
même communauté.
est la proportion des longueurs d’arêtes monochromes
dans le chemin k entre les nœuds i et j appartenant à la
même communauté.
La distance Spinp
(«Shortest path internal node
proportion») est la proportion des noeuds internes
d’un chemin le plus court appartenant à la même communauté d’espèces à ces deux
extrémités (Formule 6). Si le chemin le plus courts ne contient pas de noeuds internes, la longueur est donc de deux, la valeur 1
est ajouté au numérateur et au dénominateur.
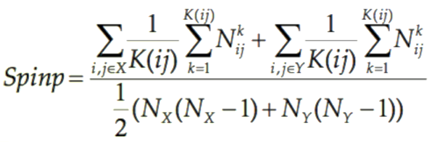
Formule 6: Formule de distance d’évolution Spinp
où Spinp est la distance d’évolution Spinp, x et y sont deux communauté d’espèce, K(i,j) est le nombre de chemins entre les nœuds i et j, NX
et NY sont le nombre de nœuds dans les communautés X et Y. ![]() est la proportion des nœuds internes de la même
communauté dans le chemin k entre i et j qui appartiennent
à la même communauté d'espèces.
est la proportion des nœuds internes de la même
communauté dans le chemin k entre i et j qui appartiennent
à la même communauté d'espèces.
Virus de la grippe de type A
Les virus de la grippe de type A ont une
génétique très diversifié du à deux phénomène: la dérive antigénique et le
réassortiment.17 La dérive antigénique découle de l’ARN polymérase
des virus de la grippe qui ont une faible fidélité induisant ainsi un haut taux
d’erreur de réplication.17 Le réassortiment survient lorsqu’une
cellule infectée par deux virus de la grippe différents et que ces virus
s’échangent des segments de gènes lors de leurs réplications.18
Les nouveaux virus ainsi formés possèdent des caractéristiques hydrides des
virus parentaux. Ces deux phénomènes ont mené à l'émergence de nouvelle souches
de virus de la grippe porcine AH1N1 qui a causé la première pandémie de grippe
du 21e siècle.
Le mode de transmission du virus de la
grippe A requiert l’activité protéique de ses deux glycoprotéines de surface,
l’hémagglutinine (HA) et la neuraminidase (NA).19 La HA permet
la fusion de la membrane virale à la membrane intracellulaire endosomale en se
liant aux récepteur de l’acide sialique. La NA clive ces récepteurs afin de
réduire l’affinité de d’autres virus envers la même cellule hôte.19
Dû à sa fonction essentielle dans l’efficacité de virulence et sa position en
surface du virus, la glycoprotéine HA est au cœur de la stratégie immunitaire
de l’hôte.19 Ainsi, un taux de mutation élevé de la structure
protéique est nécessaire afin d’échapper au système immunitaire de l’hôte.
En se basant sur ces connaissances, nous
avons voulu comparer l’évolution et la diversification des séquences HA des
virus AH1N1 infectant l’humain et les porc. Pour ce
faire, nous avons d’abord récupérer les séquences des HA des virus AH1N1
provenant soit de l’humain ou du porc. À partir de l'alignement multiples de
ces séquences, nous avons créer un arbre phylogénétique et un réseau de
similarité sur lequel nous avons mesuré les cinq distances évolutives développé
par le groupe du Dr Makarenkov.
MATÉRIELS ET MÉTHODES
Élaboration du jeu de données
Cinq séquences humaines (Numéro
d'accession : ACQ89903, ACY78004, ACY77904, AFY24010, ACY77944) et sept
porcines (Numéro d'accession NCBI : AKJ82484, ADG58953, ADU02154, ACY82404,
ADA63254, ADG01948, ADU02153) de la protéine HA prélevées et séquencées lors de
la pandémie de AH1N1 non-identique ont été sélectionnées aléatoirement sur la
base de données du virus de l’influenza des serveurs du NCBI.20
Toutes les séquences humaines et porcine proviennent respectivement du Mexique
et de l’Asie ainsi que l’Europe.
Réseau phylogénétique
Un alignement multiple des séquences a été
effectué avec l’outils MUSCLE.21 À l’aide du programme OmicsBox (OmicsBox, 2019) une
base de données local de BLAST contenant uniquement les 12 séquences a été
créer. Les pourcentages de similarité ont été obtenus par un BLASTp entre les séquences contre la base de donnés locale.
À partir de ces pourcentages de similarité, un réseau igraph
dans R a été produit.
Arbre phylogénétique
À partir de l’alignement multiples de
MUSCLE, un arbre phylogénétique a été construit et visualisé avec l’outil phyML dont le modèle utilisé est Flu
+ G.21,22
Distances évolutives
La distance UniFrac a été calculés selon
la Formule 1. Les distances évolutives ont été calculés selon la documentation
de l’application R NetFrac.
RÉSULTATS
L’arbre phylogénétique HA de la Figure 3
contient 12 séquences de la protéine HA de 566 acide aminées. Nous avons utilisé
le modèle de substitution d’acide aminé FLU, puisque celui-ci a été conçu pour
estimer la distance des séquences provenant de la grippe. Le taux de variation
G considère l’hétérogénéité du taux de substitution entre les sites selon une
distribution gamma. Nous avons utiliser la distance NNI «Nearest Neighbor
Interchange » et la distance SPR «Subtree pruning and regrafting» dans le
but d’obtenir le meilleur embranchement possible. Avec l’une ou l’autre des
méthodes les arbres résultants sont ressemblant et toutes les branches ont une
longueur de zéro. De ce fait, la distance évolutive UniFrac est elle-même de
zéro.
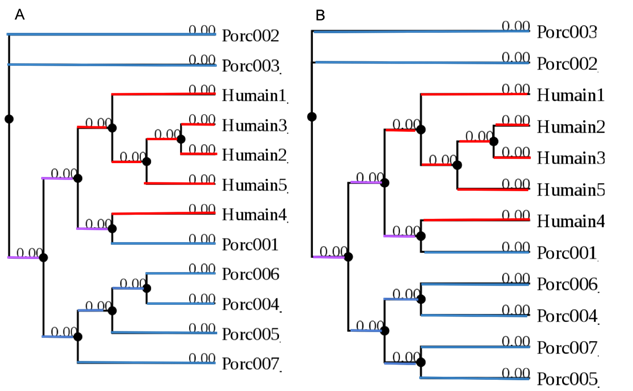
Figure
3 : Arbres phylogénétique HA du virus de la grippe porcine AH1N1 obtenue A avec
la distance NNI et B avec la distance SPR. Toutes les séquences humaines
proviennent du Mexique. Les séquences porc001, porc003, porc004, porc005,
porc006, porc007 proviennent respectivement du Royaume-Unis, Corée du Sud,
Singapour, Taiwan et de la Thaïlande. Les séquences porc002 et porc003 de la
Chine.
Le réseau de la Figure 4 exprime les
relations de similarités des séquences protéiques et est composé de 12 nœuds et
20 arêtes. Chaque séquence protéique étudiée représente un nœud du réseau et
chaque arête correspond à un taux de similarité d’au moins 99.7% entre deux
séquences. Chaque arête est accompagnée d’un poids relatif à la proximité des
deux séquences et d’un multiplicateur de 1,5 fois si les deux séquences
appartiennent à la même espèce, afin de simuler la facilité relative de
transmission. On peut observer deux séquences centrales au réseau (Porc7 et
Humain2) qui ont chacun neuf arêtes. Seule le nœud Humain5 ne possède aucune
arête monochrome, celle-ci n’étant relié qu’à une seule séquence porcine.
Également, le nœud Porc2 est la seule séquence qui est uniquement reliée par
une arête monochrome.
![]()
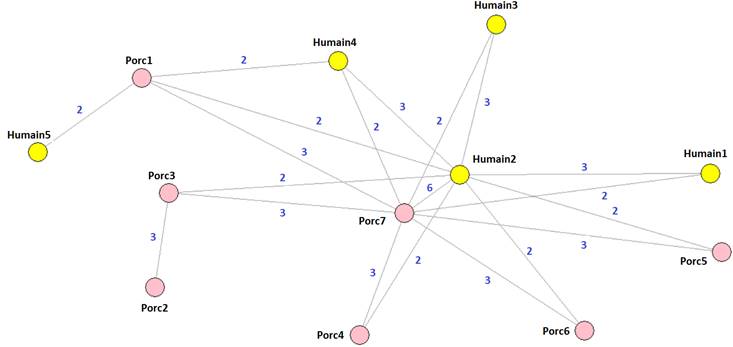
Figure 4 : Réseau phylogénétique de similarité de séquences
protéiques d’hémagglutinine provenant de la grippe AH1N1 humaine et porcine.
Les arêtes représentent un pourcentage de similarité entre deux séquences est
supérieur à 99,7%. Les séquences humaines et porcines sont représentées
respectivement par les nœuds jaunes et roses. Les séquences porc001, porc003,
porc004, porc005, porc006, porc007 proviennent respectivement du Royaume-Unis,
Corée du Sud, Singapour, Taiwan et de la Thaïlande. Les séquences porc002 et
porc003 de la Chine.
Les résultats des mesures de distances,
tels que représentés dans le tableau I, ont été obtenus à partir de
l’application R NetFrac, lorsqu’appliqué sur le réseau phylogénétique des
séquences de la grippe AH1N1. On peut y observer des valeurs relativement
faibles de distances Spp «shortest path proportion»,
Spep «shortest path edge proportion», Spelp «shortest path edge length
proportion» ainsi que la distance de transfert. La distance NetUniFrac de 0,51
indique un partage presque uniforme des séquences entre les deux souches. Le
résultat de la distance Spinp «shortest path internal node proportion» est
nettement plus élevé que les autres distances, avec un valeur de 0,79.
Tableau
I : Résultats des mesures de distances évolutives offertes par le programme
NetFrac, lorsqu’appliqué au réseau phylogénétique de la Figure 4.
|
Distance |
Résultat |
|
NetUniFrac |
0.51 |
|
Spp |
0.32 |
|
Spep |
0.39 |
|
Spelp |
0.41 |
|
Spinp |
0.79 |
|
Transfert humain -
porc |
0.30 |
|
Transfert
inverse porc -
humain |
0.33 |
DISCUSSION
Les processus de virulence de la grippe A
et les réponses immunitaires de l’hôte sont à la source du haut taux de
mutations observées dans le génome viral du AH1N1. Parfois, des sauts d’espèces
hôtes peuvent se produire suite à ces évènements de mutations, comme les
événements de la pandémie de 2009 le démontre. La protéine HA se trouve au cœur
de ces mutations et du processus de virulence dû à son rôle dans
l’internalisation du virion et le ciblage qu’en fait le système immunitaire.
Ainsi, l’évolution entre les HA provenant de l’humain et du porc a été le sujet
d’étude. Pour ce faire, un jeu de donnée contenant 12 séquences HA dont cinq
provient de l’humain et sept provient du porc a été créé. À partir de
l’alignement de ces séquences, un arbre ainsi qu’un réseau phylogénétique ont
été construit.
La distance UniFrac calculée pour l’arbre
phylogénétique de la Figure 3 a une valeur de zéro. Ceci découle du fait que
toutes les branches de l’arbre HA avait une longueur de zéro. La longueur des
branches d’un arbre est proportionnelle à la distance évolutive entre les deux
séquences. Le tout indique que toutes les séquences sont identiques. Or,
l’alignement multiples des séquences prouve que celle-ci ne sont pas identiques
à 100%. La résolution de phyML de 10-2 est
trop faible afin de détecter le peu de mutations entre les séquences étudiées.23
À partir de l’alignement multiples des séquences (Figure supp 1 et 2), il
est possible de calculer (1 mutation / 566 a.a) que
la résolution minimale est de 10-3, puisqu’il y a au minimum il y a
une mutation pour 566 site.
La distance NetUniFrac, qui est une
généralisation de la distance UniFrac pour les réseaux, est basée sur la
proportion du nombre d’arêtes monochrome du réseau par rapport au nombre total
d’arêtes. La valeur de 0,51 obtenue signifie que la moitié des arêtes sont monochromes.
Cela concorde avec l’observation visuelle du réseau. Les deux séquences
centrales (Humain2 et Porc7) sont reliées à la grande majorité des autres
séquences, la moitié étant monochrome.
Les mesures Spp,
Spep et Spelp calculés sur
le réseau sont toutes sensibles au plus court chemin monochrome entre deux
points. Cependant, la présence des deux nœuds centraux connectant la grande
majorité des autres séquences affecte l’application de ces mesures, qui sont
respectivement 0,32, 0,39 et 0,41. Ces valeurs sont rapprochées et démontrent
que dans ce type de réseau, les variations du calcul du plus court chemin (le
nombre d’arêtes dans le cas de Spep et leur poids
dans le cas de Spelp) n’ont pas une incidence marquée
sur la valeur finale obtenue. Ainsi, ces variations du calcul de mesure
suggèrent que les séquences humaines et porcines sont relativement similaires.
Cela concorde avec la théorie, considérant que le seuil de sélection des
séquences était de 99,7% de similarité.
Cependant, la valeur Spinp,
qui prend en compte les nœuds internes monochrome ainsi que la pertinence
statistique des espèces associées aux nœuds a produit une valeur de 0,79, ce
qui est fortement supérieur aux autres variations de la mesure du plus court chemin
et suggère que les séquences humaines et porcines sont relativement moins
similaires.
De plus, les résultats de transfert et
transfert inverse, qui représentent la proportion des chemins les plus courts
étant monochrome, ont été déterminé comme étant similaire entre l’humain et le
porc (0,30 et 0,33). Cette valeur signifie en théorie que beaucoup de gènes ont
été transmis d’une espèce vers l’autre et vis-versa. Il est probable que le
haut taux de mutation de la protéine HA mène à des représentations similaires
chez l’humain et le porc de façon indépendante. Dans ce cas-ci, les valeurs de
transfert auraient donc un pouvoir de prédiction limité.
CONCLUSION
En conclusion, des séquences virales de
AH1N1 porcines et humaines très similaires ont été utilisées pour produire un
arbre et un réseau phylogénétique. Il n’a pas été possible de détecter de
différence évolutive à partir de l’arbre phylogénétique. Le réseau a permis
obtenir divers mesure de distances évolutive entre les communautés à l’aide du
programme NetFrac. Tel qu’attendu, les trois mesures Spp,
Spep et Spelp obtenues
indique que nos échantillons étaient similaires. Cependant, la mesure Spinp, basée sur le calcul des nœuds interne des plus
courts chemins a produit une valeur beaucoup plus forte, qui suggère une
similarité plus faible entre les communautés. Le tout tant à démontrer que les
réseaux phylogénétiques et les nouveaux calculs de distance développée par
l’équipe du Dr Makarenkov sont plus adaptés afin de représenter les phénomènes
évolutifs complexes, tel que le réassortiment.
Il serait intéressant de tester
l’application NetFrac avec des réseaux viraux de taille plus grande afin de
produire des résultats avec plus de poids statistique et moins de biais de
sélection. De plus, les mesures de plus court chemins pourraient être testés
avec des réseaux reflétant divers degrés de séparation entre les communautés
afin de voir le comportement de chaque variation de la mesure Spp.
RÉFÉRENCES
1. Doolittle WF, Brunet TD. What Is the Tree of Life? PLoS Genet. 2016 Apr 14;12(4):e1005912.
2. Doolittle WF, Bapteste E. Pattern pluralism and the tree of life
hypothesis. Proc Nat Acad Sci USA.
2007;104(7):2043–9.
3. Merico D, Gfeller D, Bader GD. How to visually interpret biological
data using networks. Nat Biotechnol.
2009;27(10):921-4.
4. Gogarten JP, Townsend JP. Horizontal gene transfer, genome
innovation and evolution. Nat Rev Microbiol. 2005;3(9):679–87.
5. Soucy SM, Huang
J, Gogarten JP. Horizontal gene transfer: building
the web of life. Nat Rev Genet. 2015;16(8):472–82.
6. Mallet J.
Hybrid speciation. Nature. 2007;446(7133):279–83.
7. Fontaine MC,
Pease JB, Steele A, Waterhouse RM, Neafsey DE et al.
Extensive introgression in a malaria vector species complex revealed by
phylogenomics. Science. 2015;347(6217):27–8.
8. Martin DP, Lemey P, Posada D. Analysing recombination in nucleotide
sequences. Mol Ecol Resour.
2011;11(6):943–55.
9. Bapteste E, van Iersel L, Janke
A, Kelchner S, Kelk S, et
al. Networks: expanding evolutionary thinking. Trends Genet. 2013;29(8):439–41.
10. Huson DH, Bryant D. Application of phylogenetic networks in
evolutionary studies. Mol Biol Evol.
2006;23(2):254–67.
11. Huson DH, Rupp R, Scornavacca C.
Phylogenetic Networks: Concepts, Algorithms and Applications. Cambridge: Cambridge
University Press; 2010.
12. Morrison DA.
Introduction to Phylogenetic Networks. Uppsala: RJR Productions; 2011.
13. Gusfield D.
ReCombinatorics: The Algorithmics of Ancestral Recombination Graphs and Explicit
Phylogenetic Networks. Cambridge: MIT Press; 2014.
14. Boucher B,
Jenna S. Genetic interaction networks: better understand to better predict.
Front Genet. 2013;4:290.
15. Lozupone C,
Knight R. UniFrac: a new phylogenetic method for comparing microbial
communities. Appl Environ Microbiol. 2005 Dec;71(12):8228-35.
16. Miryala SK,
Anbarasu A, Ramaiah S. Discerning molecular interactions: A comprehensive
review on biomolecular interaction databases and network analysis tools. Gene.
2018;642:84-94.
17. Mondal SI, Zubaer A, Thapa S, Saha C, Alum MA, et al. Envelope Proteins Pertain with Evolution and Adaptive Mechanism of the Novel Influenza A/H1N1 in Humans. J Microbiol Biotechnol. 2010 Nov;20(11):1500-5.
18. Bao Y, Bolotov
P, Dernovoy D, Kiryutin B, Zaslavsky L, et al. The influenza virus resource at
the National Center for Biotechnology Information. J Virol. 2008
Jan;82(2):596-601.
19. Prachanronarong KL, Canale AS, Liu P, Somasundaran M, Hou S, et al. Mutations in Influenza A Virus Neuraminidase and Hemagglutinin Confer Resistance against a Broadly Neutralizing Hemagglutinin Stem Antibody. J Virol. 2019 Jan 4;93(2)
20. Bao Y, Bolotov P, Dernovoy D, Kiryutin B,
Zaslavsky L, et al. The influenza virus resource at the National Center for
Biotechnology Information. J Virol. 2008 Jan;82(2):596-601
21. Madeira F, Madhusoodanan N, Lee J, Tivey ARN, Lopez R. Using EMBL-EBI Services via Web Interface and Programmatically via Web Services. Curr Protoc Bioinformatics. 2019 Jun;66(1):e74.
22. Guindon S,
Dufayard JF, Lefort V, Anisimova M, Hordijk W, et al. New algorithms and
methods to estimate maximum-likelihood phylogenies: assessing the performance
of PhyML 3.0. Syst Biol. 2010 May;59(3):307-21.
23. Wong RG, Wu JR, Gloor GB. Expanding the UniFrac Toolbox. PLoS One. 2016 Sep 15;11(9):e0161196.
ANNEXE

Figure Supp 1 : Alignement multiple
des position 1 à 300 des séquences HA du virus
AH1N1.

Figure Supp 2 : Alignement multiple
des position 301 à 566 des séquences HA
du virus AH1N1.