Algorithmes bioinformatiques pour
la reconstruction d’arbres consensus et de super-arbres multiples
Table des matières
1.
La phylogénie et l’analyse d’inférence d’arbre phylogénétique
Distance
des moindres carrés (LS)
Distance de Robinson et Foulds (RF)
Dissimilarité
de bipartitions (DB)
Validation
de l’approche et des critères
Fonctions
objectives W’ et W’’
Validation
de l’approche et des critères
I. Introduction
1. La phylogénie et l’analyse d’inférence d’arbre phylogénétique
La phylogénie est l’étude des relations de parenté entre les
organismes. Les buts de la phylogénie sont, entre autres, d’étudier l’évolution
des êtres vivants et d’estimer des temps de divergence depuis leur dernier
ancêtre commun. Les étapes d’une analyse phylogénétique
de données moléculaire consistent à obtenir les séquences des gènes (extraction
génomique => PCR => séquençage), de les aligner, de calculer les
distances ou les similarités entre les espèces (selon la méthode d’inférence
choisie) et d’appliquer une méthode de reconstruction d’arbre. Une fois l’arbre
phylogénétique obtenu, il faut pouvoir comparer les arbres puisque les
différentes méthodes de reconstruction d’arbres donnent des résultats
différents. De plus, selon les informations choisies pour effectuer l’analyse,
les phylogénies diffèrent, car chaque gène possède sa propre histoire
évolutive.
2. Comparaison d’arbres
Des métriques existent pour comparer
objectivement les arbres. Les principales mesures de comparaison d’arbres sont
les suivantes :
-
Distance
des moindres carrés (LS)
-
Distance
de Robinson et Foulds (RF)
-
Distance
des quartets (QD)
-
Dissimilarité
de bipartitions (DB)
Distance des moindres carrés (LS)
La
distance des moindres carrés (Gauss, 1975) est la somme des différences entre la valeur observée
et la valeur attendue au carrée. Il est souhaitable d’obtenir la valeur
minimale. Son équation est illustrée ci-dessous :
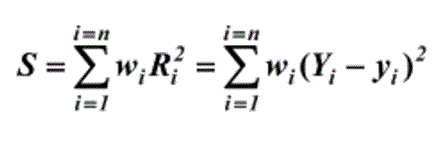
Où S = Somme
R = Résidu (différence entre la valeur observée et la valeur attendue)
(1)
Équation de la distance des moindres
carrés (Tirée de Wolberg, 2006)
Distance de Robinson et Foulds (RF)
La
distance de Robinson et Foulds (Robinson et Foulds, 1981), aussi connu sous le nom
de différence symétrique, est une mesure de distance topologique entre 2
arbres. C’est le nombre minimum d’opérations élémentaires ( ![]() et
et ![]() ,
fusion et séparation de nœuds respectivement) pour passer d’un arbre T à un
arbre T’ (Robinson et Foulds, 1981). La distance RF peut être également
vue comme le nombre de bipartitions non partagées entre les 2 arbres
(Felsenstein, 2004). Cette métrique
prend la valeur 0 lorsque les deux arbres sont pareils. La figure 1 illustre un
exemple de la distance de RF.
,
fusion et séparation de nœuds respectivement) pour passer d’un arbre T à un
arbre T’ (Robinson et Foulds, 1981). La distance RF peut être également
vue comme le nombre de bipartitions non partagées entre les 2 arbres
(Felsenstein, 2004). Cette métrique
prend la valeur 0 lorsque les deux arbres sont pareils. La figure 1 illustre un
exemple de la distance de RF.
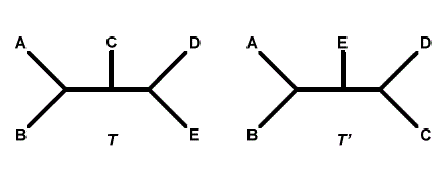
Fig 1. La distance RF entre les deux arbres T et T’ est d(T, T’)=2, soit la séparation du nœud
entre D et E et la fusion de C et D. (Tirée de Tahiri, 2015)
Distance des quartets (QD)
La
distance des quartets (Bryant et al.,
2000) est le nombre de quadruplets (sous-arbre généré par 4 feuilles) qui ont
une topologie différente dans les deux arbres considérés. La figure 2 illustre
les différentes typologies que peut prendre un quadruplet.
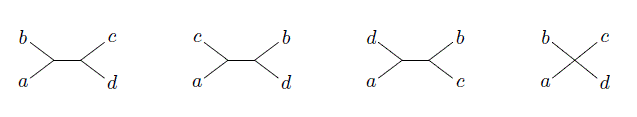
Fig 2. Les 4
topologies possibles du quadruplet formé par les espèces a, b, c et d (Tiré de
Brodal et al., 2001).
Dissimilarité de bipartitions (DB)
La dissimilarité de bipartitions
(Boc et al., 2010, Makarenkov et al., 2007) peut être considérée comme
une mesure plus fine de la distance RF étant donné que cette dernière ne
considère que les bipartitions identiques. Pour deux arbres ayant le même
nombre de feuilles, on construit une table de bipartitions pour chaque arbre,
chacune contenant les vecteurs de bipartition associés aux branches internes de
l’arbre (0 pour les feuilles d’un côté et 1 pour les feuilles de l’autre côté)
(Fig 3).
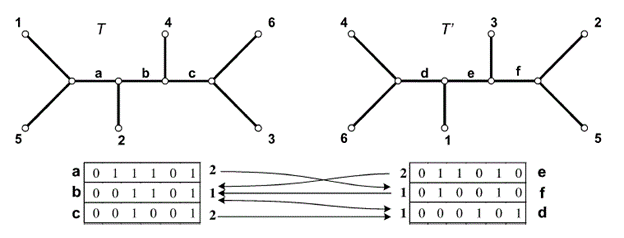
Fig 3. Deux
arbres, T et T’, et leur table de
bipartitions. Chaque ligne de la table de bipartition correspond à une branche
interne. Les flèches indiquent les associations entre les vecteurs de
bipartitions des 2 tables. Les valeurs en gras proches de chaque vecteur
représentent la distance qui y est associé. (Traduit et tirée de Boc et al., 2010).
La
distance bd est calculée comme
suit :
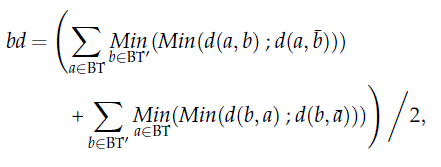
Où BT est la
table de bipartitions de l’arbre T ;
BT’ est la table de bipartitions de
l’arbre T’ ;
d(a, b) est la distance de Hamming entre
le vecteur a et b ;
ā et ƀ sont les compléments des
vecteurs a et b.
(2)
Équation de la dissimilarité de bipartitions (Tirée de Boc et al., 2010)
Au-delà
de la comparaison d’arbres, il faut également pouvoir retirer des informations
des différents arbres obtenus. Plusieurs approches existent soit pour garder ce
qui est commun aux arbres, les méthodes d’arbres consensus, soit pour fusionner
les arbres en un seul, les méthodes de super-arbres. En effet, pour des projets
tels que le Tree of Life (Madison, 2007), il est nécessaire de pouvoir
fusionner des arbres ensemble et de retirer ce qui leur est commun.
II. Arbres de consensus
1. Méthodes principales
Les méthodes principales
d’inférence d’arbres consensus sont les méthodes de consensus strict, de
consensus majoritaire et de consensus majoritaire étendu (Fig 4). La méthode de
consensus strict (Sokal et Rohlf, 1981) ne garde que les bipartitions présentes
dans tous les arbres. Ainsi, seules les relations entre 2 taxons retrouvées
dans tous les arbres sont conservées. La méthode du consensus majoritaire
(Margush et McMorris, 1981) ne retient que les bipartitions présentes dans 50%
(ou un autre pourcentage spécifié) des arbres. La méthode de consensus
majoritaire étendu (Felsenstein, 1985), aussi connu sous le nom de
« greedy consensus tree method », conserve les bipartitions de
manière séquentielle. Les bipartitions sont ordonnées par ordre décroissant de
fréquence d’occurrence. En suivant cet ordre, on choisit la bipartition la plus
fréquente et on rajoute les autres bipartitions, une à une, selon leur
compatibilité avec les bipartitions déjà choisies (Bryant et al., 2003).
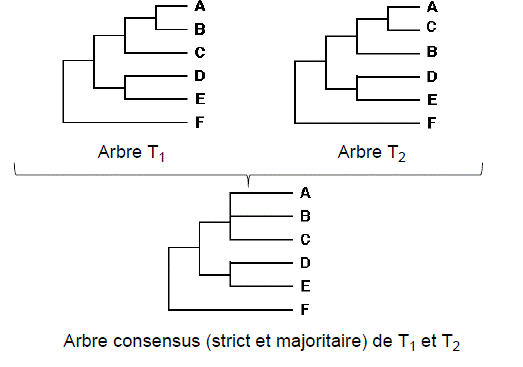
Fig
4. Dans le cas d’un ensemble de 2 arbres, l’arbre de consensus strict est le
même que l’arbre de consensus majoritaire (Tirée de Tahiri, 2015).
Les
méthodes précédentes ont en commun qu’elles ne permettent d’obtenir qu’un seul
arbre consensus. Ce n’est pas toujours idéal, car il y a souvent perte
d’informations. Par exemple, dans la figure 5, l’approche classique d’arbre
consensus majoritaire unique nous retourne une arborescence étoilée non informative
alors que si on permettait des arbres consensus majoritaires multiples, on
conserverait des informations potentiellement intéressantes.
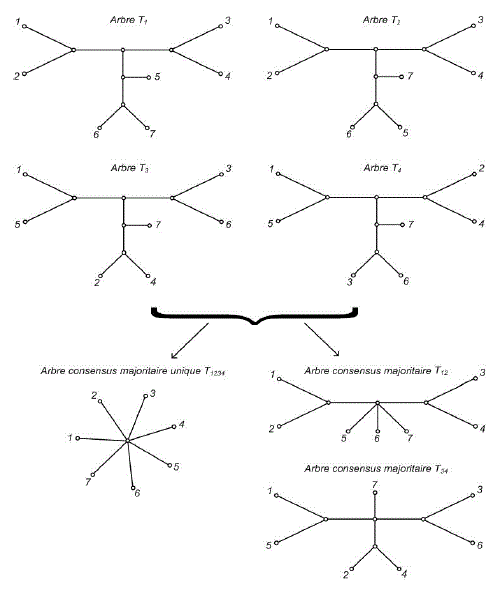
Fig 5. Quatre
arbres phylogénétiques T1, T2, T3 et T4 définis sur un ensemble de sept feuilles
; leur arbre consensus majoritaire unique T1234; la solution à deux arbres
consensus majoritaires T12 et T34 (Tirée de Tahiri et al., 2014).
L’étude
de Tahiri et al. (2014 et présentement) propose un nouvel algorithme
consensus-trees qui, à partir d’un ensemble d’arbres, permet d’obtenir un ou
plusieurs arbres consensus en regroupant les arbres phylogénétiques.
2. Algorithme consensus-trees
L’algorithme consensus-trees est
basé sur l’idée que classifier les arbres phylogénétiques avec l’algorithme des
k-moyennes permettrait de séparer les familles de gènes avec la même histoire
évolutive. Cet algorithme est basé sur l’algorithme des k-moyennes afin
d’obtenir des partitions optimaux d’arbres. La distance de Robinson et Foulds a
été utilisée et les critères d’évaluation CH (Calinski-Harabasz, 1974) et W
(nouvelle fonction objective développée). La propriété utilisée est que selon
la distance de Robinson et Foulds, l’arbre médian au sein d’un groupe d’arbres
est l’arbre consensus (Barthélemy et McMorris, 1986).
Les données en entrée prises par
l’algorithme sont n arbres
phylogénétiques définis sur un même ensemble d’espèces (taxons). Tous les
arbres doivent donc avoir les mêmes feuilles. Les données en sortie sont (1) le
partitionnement optimal de l’ensemble d’arbres en un ou plusieurs groupes et
(2) pour chaque groupe, (a) la liste des arbres qui s’y retrouvent; (b) l’arbre
consensus du groupe; (c) la valeur des indices de CH et W.
Algorithme des k-moyennes
L’algorithme
des k-moyennes partitionne de manière optimale un ensemble de données en k groupes selon un critère de similarité
(MacQueen, 1968). Les étapes de
l’algorithme standard sont les suivantes : (1) un nombre k de « moyennes » sont
choisies aléatoirement parmi les éléments (i.e. : les données); (2) des
groupes sont créés en associant chaque élément à la « moyenne » la
plus proche selon une distance choisie; (3) le centroïde de chaque groupe est
calculé et il devient la nouvelle « moyenne »; (4) les étapes 2 et 3 sont répétées jusqu’il
y a convergence. Déterminer le nombre k de groupes optimal est souvent
difficile. L’évaluation du k peut se
faire après l’analyse avec un critère d’évaluation.
Propriété
Cette
propriété a été décrite par Barthelemy et McMorris (1986). L’arbre médian d’un
regroupement d’arbres est celui dont la distance totale avec tous les autres
arbres de ce groupe est la plus petite possible. Dans le cas où cette distance
est la distance RF, cet arbre médian correspond à l’arbre de consensus
majoritaire lorsqu’il y a un nombre impair d’arbres dans ce groupe. Lorsque le
nombre d’arbres dans le groupe est pair, il est possible d’avoir 2 arbres
« médians » (Felsenstein, 2004).
La
variante heuristique de l’algorithme des k-moyennes sur lequel est basé
l’algorithme consensus-tree est celle implémentée par Makarenkov et Legendre
(2001). La métrique choisie est la distance de Robinson et Foulds et les
critères d’évaluation pour déterminer le k optimal sont CH et W.
Indice de CH
L’indice
de Calinski-Harabasz (1974) permet de déterminer le nombre de groupe optimal
pour, entre autres, l’algorithme des k-moyennes. Plus la valeur de l’indice est
élevée, meilleur est le partitionnement des groupes. L’équation de l’indice CH
est donnée ci-dessous :

(3)
Équation de Calinkski-Harabasz (Tiré de Tahiri, 2015)

(4)
Équation de l’indice d’évaluation intergroupe (Tirée de Tahiri, 2015)

(5)
Équation de l’indice d’évaluation intragroupe
(Tirée de Tahiri, 2015)
La
limite de l’indice de CH est qu’il ne permet pas de déterminer le cas où k=1
(un seul groupe=> arbre consensus unique). Il n’est donc pas possible de
comparer la situation de k=1 et les situations de k>2 à k=n-1 (plusieurs
groupes dont au moins un groupe contenant
≥ 2 éléments => arbres consensus multiples).
Indice W (fonction objective)

(6) Équation de la fonction objective W (Tirée de
Tahiri, 2015)
La
nouvelle fonction objective W, dont l’équation se retrouve ci-haut, permet de
calculer le cas d’un arbre consensus unique (k=1), mais il y a une perte
d’informations concernant les distances intergroupes, puisque cette fonction ne
prend en compte que la distance intragroupe.
Validation de l’approche et des critères
Des simulations seront effectuées
afin de valider l’approche et le choix des critères. Le plan des simulations
comprend 3 étapes :
(1) Générer
k arbres phylogénétiques binaires
aléatoires à n feuilles avec le site T-Rex (Boc et al., 2012) avec des paramètres de k={1…10} et n= {9,16,32,64).
(2) Générer
pour chaque arbre Ti (où
i=1…k), un ensemble de 100 arbres aléatoires appartenant à la classe
i et ce, pour chaque intervalle suivant :
Intervalle
I (0 à 10% de similitude);
Intervalle
II (10 à 25% de similitude);
Intervalle
III (25 à 50% de similitude);
Intervalle
IV (50 à 75% de similitude);
Les
pourcentages de similitude sont mesurés avec la distance RF.
(3) Appliquer
l’algorithme consensus-trees aux ensembles d’arbres générés avec les différents
paramètres (k, n, Intervalle, Fonction Objective W et le critère CH).
Résultats
de l’étude préliminaire
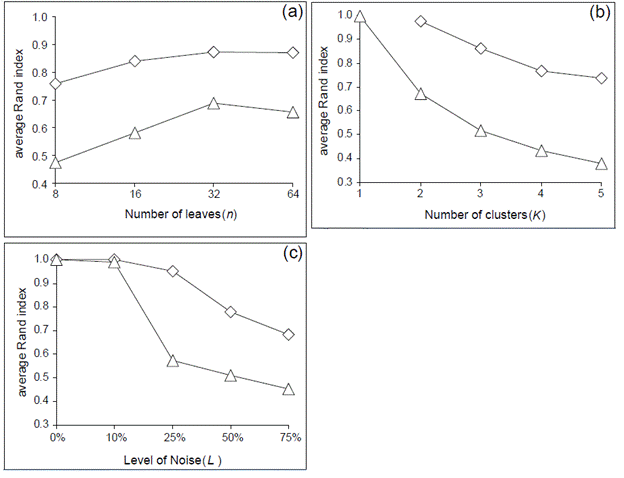
Fig 6. Étude préliminaire de l’évolution
de l’indice Rand moyen (a) en
fonction du nombre de feuilles (n); (b) en fonction du nombre de partitions
(k); (c) en fonction du pourcentage de similitudes (L) entre les arbres phylogénétiques (Intervalle) pour les deux
critères :
◊ Calinski-Harabasz; ∆ W
(Tiré de Tahiri, 2015).
Pour
les simulations, on connaît k, le
nombre de groupes susceptibles d’être retrouvé par l’algorithme des k-moyennes.
On sait également que les arbres appartenant aux intervalles plus bas (e.g.
intervalle I) sont les arbres les plus éloignées. Pour connaître la force de
l’algorithme, il faut donc regarder si les arbres éloignés sont regroupés
ensemble ou non. L’indice de Rand est critère de fiabilité des groupes. Les
résultats de l’étude préliminaire suggèrent qu’il est plus difficile de
partitionner les arbres lorsque le nombre n de feuilles est élevé (Fig 6a). De
même, plus il y a de groupes k, moins ils sont mis ensemble (Fig 6b). Plus les
arbres sont éloignées (0 à 10%), plus il est difficile de bien les partitionner
(Fig 6c).
En
somme, l’algorithme consensus-trees est une méthode de partitionnement d’arbres
phylogénétiques qui permet d’inférer des arbres consensus unique ou multiples
selon le partitionnement obtenu. Il peut être appliqué autant pour la méthode
de consensus strict, que les méthodes de consensus majoritaire ou consensus
majoritaire étendu (Tahiri et al.,
2014).
III. Super-arbre
1. Méthodes principales
La
méthode des super-arbres permet de fusionner des arbres phylogénétiques
d’espèces différentes (arbres sources) en un seul grand arbre contenant toutes
ces espèces et le plus cohérent avec les arbres sources. Il faut par contre
qu’il y a un chevauchement entre les feuilles des arbres sources. Cette méthode
est particulièrement intéressante pour les jeux de données énormes; les
analyses peuvent être faites sur des sous-ensembles de données et mises en
commun par après. Plusieurs méthodes d’inférence de super-arbres existent dont
la méthode de Gordon (1986) utilisé dans le passé, mais menant parfois à des
incohérences. Présentement, une des méthodes les plus populaires est le MRP
(Ragan, 1992; Doyle, 1992; Baum, 1992). La méthode du MRP (Fig 7) consiste à construire
une matrice avec les arbres sources (1 pour les espèces partageant un nœud
commun dans les arbres; 0 pour les autres espèces et ? pour les espèces non
présentes) pour ensuite reconstruire l’arbre le plus parcimonieux (Von
Haeseler, 2012).
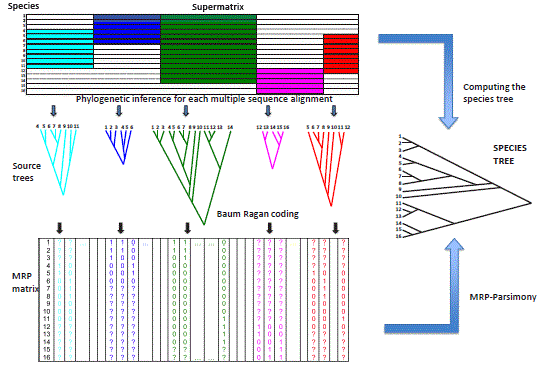
Fig 7. Étapes de la méthode MRP et de la méthode de supermatrice. La
méthode MRP encode les données des arbres sources dans une matrice MRP avant
d’appliquer une reconstruction de parcimonie pour obtenir le super-arbre
d’espèces. La méthode de supermatrice prend la supermatrice de gènes concatenés
pour obtenir l’arbre d’espèces (Tirée de
Von Haeseler, 2012).
Les
problèmes soulevés par les super-arbres sont similaires à ceux des arbres
consensus : la nécessité de fusionner des arbres phylogénétiques pour des projets
tels que le Tree of Life, l’incohérence de fusionner des arbres phylogénétiques
et la perte d’informations et la perte d’informations et le mauvais recoupement
des feuilles.
2. Algorithme Super-trees
L’algorithme
super-arbre est une généralisation de l’algorithme précédent : au lieu de
s’appliquer sur des arbres ayant les mêmes feuilles, il s’applique sur des
arbres phylogénétiques partageant partiellement les mêmes feuilles.
L’algorithme est également basé sur l’algorithme des k-moyennes.
Les données en entrées prise par
l’algorithme sont n arbres
phylogénétiques définis sur des ensembles d’espèces (taxons) différents, mais
chevauchants. La particularité est qu’il faut filtrer les ensembles d’espèces
et la difficulté réside en la détermination d’un seuil minimum de feuilles
chevauchantes entre les arbres. Les données en sortie sont (1) le
partitionnement optimal de l’ensemble d’arbres en un ou plusieurs groupes et
(2) pour chaque groupe, (a) la liste des arbres qui s’y retrouvent; (b) le
super-arbre du groupe; (c) la valeur des indices de W’ et W’’.
Fonctions objectives W’ et W’’
Les fonctions objectives W’ et W’’
sont utilisés comme indices dans l’algorithme Super-trees. Les équations de W’
et W’’ sont les suivantes :

(7)
Équation de la fonction objective W’ (Tiré de Tahiri, 2015)
La
fonction W’ est une variante de la fonction objective W. Pour chaque cas, il
faut inférer le centroïde (super-arbre majoritaire), qu’il soit réel ou
virtuel, ce qui devient coûteux.

(8)
Équation de la fonction objective W’’ (Tiré de Tahiri, 2015)
La fonction W’’ est une autre
variante de la fonction objective W, mais pour lequel il n’est pas nécessaire
de calculer le centroïde à chaque cas.
Validation de l’approche et des critères
Le
plan des simulations à venir comprend également 3 étapes :
(1) Générer
k arbres phylogénétiques binaires aléatoires ayant n1 à n2
feuilles dont au moins n feuilles
communes avec le site T-Rex (Boc et al., 2012) avec des paramètres de k={1…10}
et n= {9,16,32,64).
(2) Générer
pour chaque arbre Ti (où
i=1…k), un ensemble de 100 arbres aléatoires appartenant à la classe
i et ce, pour chaque intervalle suivant :
Intervalle
I (0 à 10% de similitude);
Intervalle
II (10 à 25% de similitude);
Intervalle
III (25 à 50% de similitude);
Intervalle
IV (50 à 75% de similitude);
Les
pourcentages de similitude sont mesurés avec la distance RF.
(3) Appliquer
l’algorithme Super-trees aux ensembles d’arbres générés avec les différents
paramètres (k, n, n1,
n2, Intervalle, Fonction
Objective= W’’ et les critères
CH, BH, LogSS et Silhouette).
IV. Applications
L’algorithme
Consensus-tree a été appliqué pour la classification des protéines ribosomales
des archaebactéries. Le jeu de données de l’étude de Matte-Taillez et al. (2002) a été utilisé. Ces données
consistent en 49 protéines ribosomales provenant de 14 archaebactéries. Les
buts de l’application de l’algorithme Consensus-tree étaient de déterminer les
protéines qui partagent la même histoire évolutive et la détection de gènes
ayant subis les mêmes transferts horizontaux.
Les résultats de l’algorithme sont illustrés dans la figure
9. Le nombre de groupe optimal (k) déterminé par les critères CH et W sont k=2
et k=1 respectivement. Les 2 arbres consensus obtenus selon le critère de CH
sont illustrés dans la figure 10 et l’arbre consensus obtenu selon le critère
W, dans la figure 11. Le problème est qu’il y a présence de minimum local et de
maximum local. Plusieurs scénarios de transfert horizontaux de gènes ont été
également simulés selon les différentes valeurs de K, soit entre K=2 et K=5
(fig 11).
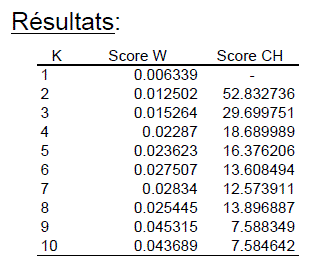
Fig 9. Résultats de
l’algorithme Consensus-tree sur les données des protéines ribosomales des
archaebactéries. Le k optimal pour le critère CH et W sont respectivement 2 et
1. (Tirée de Tahiri, 2015)
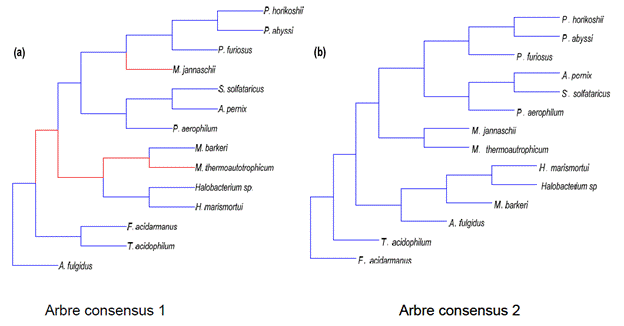
Fig 10. Arbres
consensus obtenus selon le critère CH (Tirée de Tahiri, 2015)
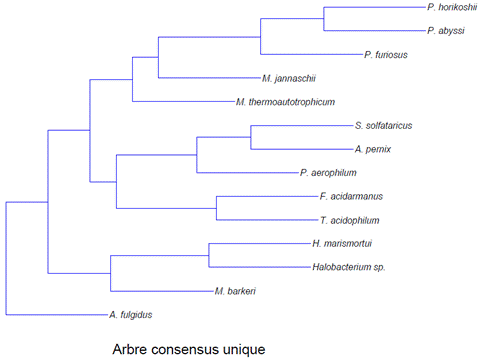
Fig 10. Arbre
consensus unique obtenu selon le critère W (Tirée de Tahiri, 2015)
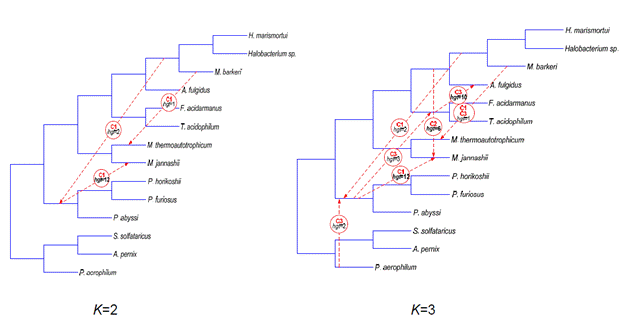
Fig 11. Scénario de
transferts horizontaux selon le k. Ici, les cas k=2 et k-3 ont été illustrés.
(Tirée de Tahiri, 2015).
Éventuellement, l’algorithme sera aussi appliqué à d’autres
jeux de données tels que les papilionidés (Gepts et al., 2005), les marsupiaux (Cardillo et al., 2004), les mammifères placentaires (Beck et al., 2006) et les oiseaux de mer
(Kennedy et Page, 2002).
Finalement, l’algorithme sera aussi appliqué pour étudier
l’évolution des langues Indo-Européennes. La base de données provient de Dyen et al. (1997) et a été améliorée par Boc
et al. (2010). Elle est regroupée en
200 mots de la liste Swadesh, traduite dans 87 langues et structurée en 1315
cognats. Le but de cette analyse est de trouver des groupes de langues
partageant la même histoire évolutive et d’appuyer soit l’hypothèse Kourgane ou
Anatolienne sur l’origine des langues indo-européenne ou peut-être émettre une
nouvelle hypothèse.
En
conclusion, les perspectives futures concernant les algorithmes proposés par
Tahiri et al. (2014 et présent) sont d’appliquer davantage ces algorithmes à de
vrais jeux de données et l’optimisation des algorithmes.
Références
Boc, A., Philippe, H., & Makarenkov, V. (2010). Inferring and
validating horizontal gene transfer events using bipartition dissimilarity. Systematic biology, syp103.
Brodal, G. S., Fagerberg, R., & Pedersen, C.
N. (2001). Computing the quartet distance between evolutionary trees in time O
(n log2 n). In Algorithms and
Computation (pp. 731-742). Springer Berlin Heidelberg.
Bryant, D. (2003). A classification of consensus
methods for phylogenetics. DIMACS series
in discrete mathematics and theoretical computer science, 61, 163-184.
Felsenstein, J.,
& Felenstein, J. (2004). Inferring phylogenies.
MacQueen, J. (1967,
June). Some methods for classification and analysis of multivariate
observations. In Proceedings
of the fifth Berkeley symposium on mathematical statistics and probability (Vol. 1, No. 14, pp. 281-297).
Maddison, D. R.,
Schulz, K. S., & Maddison, W. P. (2007). The tree of life web project. Zootaxa, 1668 (Linnaeus Tercentenary:
Progress in Invertebrate Taxonomy), 19-40.
Robinson, D., & Foulds, L. R. (1981).
Comparison of phylogenetic trees. Mathematical
Biosciences, 53(1), 131-147.
Tahiri, N., Willems, M. & Makarenkov, V. (2014). )
Classification d’arbres
phylogénétiques
basée sur l’algorithme des k-moyennes, Proceedings of SFC 2014, Rabat,
Morocco, 49-54.
Von Haeseler, A. (2012). Do we still need supertrees?. BMC biology, 10(1), 13.
Wolberg, J. (2006). Data analysis using the method of
least squares: extracting the most information from experiments. Springer Science & Business Media.
Ce
rapport a été créé suite à la conférence
de Nadia Tahiri dans le cadre du cours BIF7002 : Séminaires en
bio-informatiques le 17 février 2015 par Tissicca Hour et Chan David Puth.