Classification du virus du papillome humain
Par Mona Parizadeh et Gaudeline R. Labrosse
Travail fait dans le cadre du cours BIF7002
UQAM – Hiver 2015
La classification taxonomique
La classification taxonomique des virus est basée principalement sur les caractéristiques phénotypiques des virus : morphologie (icosaèdre, filament), caractéristiques du génome (ARN, ADN, simple brin, double brin, linéaire, circulaire), site de réplication (noyau, cytoplasme), mode de réplication, hôtes infectés, maladies provoquées, tissus ou types cellulaires infectés (peau, muqueuses, neurones), etc. etc. Deux grands cadres sont utilisés pour la classification taxonomique des virus : le comité international de la taxonomie des virus (ICTV) et la classification Baltimore [1-3].
La classification phylogénétique
La classification phylogénétique est basée sur les données de séquençage et l’utilisation des séquences génomiques pour obtenir l’histoire évolutive et les liens de parenté entre organismes afin d’identifier des ancêtres communs. Toutefois, le concept de phylogénie appliqué aux virus est plutôt controversé, à cause du débat entourant la question : un virus est-il un être vivant? Plusieurs auteurs ne s’entendent pas sur la présence de la classification des virus dans le projet « tree of life » [4-6]. Les concepts de la phylogénie permettent toutefois de construire des arbres de relations entre les virus des différentes familles et d’établir des liens de similarité entre différentes souches ou espèces de virus. L’analyse phylogénétique chez les virus est particulièrement intéressante compte tenu de l’évolution rapide des gènes viraux.
Les méthodes phylogénétiques peuvent être utilisées en épidémiologie pour obtenir de l’information concernant les patrons d’émergence des virus. Elles permettent aussi d’estimer le temps et l’origine d’apparition d’une nouvelle souche ou encore l’émergence de nouvelles recombinaisons ou réarrangements des génomes viraux. Les études phylogénétiques des virus permettent même d’établir l’évolution au sein d’une population et d’une zone géographique. La classification phylogénétique des virus permet également d’obtenir des informations pouvant être utiles au diagnostic ou pronostic de certaines infections, de même qu’au développement d’outils thérapeutiques préventifs (vaccins) ou curatifs (antiviraux) [7].
Études phylogénétiques chez les virus
L’inférence phylogénétique a notamment été utilisée avec le virus influenza pour établir l’origine génétique des pandémie de grippe chez l’humain, pour étudier l’émergence de la grippe porcine en Amérique du Nord et pour analyser la fréquence des réassortiments du génome viral [8, 9]. Pour le virus HIV, la phylogénétique a permis de mieux comprendre les origines et la propagation initiale du virus, de même que d’aider à la classification des souches HIV et à la compréhension du mécanisme de résistance aux médicaments du virus HIV [10, 11]. Pour le virus du SRAS, les études phylogénétiques ont notamment servies à caractériser l’épidémie de SRAS et à déterminer les origines zoonotiques du virus [12] (Figure 1).
La classification phylogénétique de la famille des papillomavirus (PVs) et particulièrement pour les virus papillomes infectant l’humain (HPV) est notamment utile pour cataloguer adéquatement le risque associé au développement du cancer, en identifiant les caractéristiques génétiques des virus oncogènes.
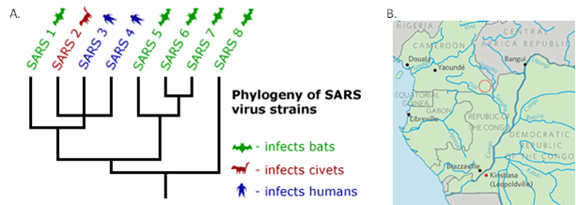
Figure 1 : Applications de la classification phylogénétique des virus. A. Étude de l’émergence zoonotique du SRAS [13]. B. Identification de l’origine de la pandémie du virus HIV-1 [14].
La Classification des papillomavirus
Le virus VPH
La famille des Papillomaviridae (PV) comprend des centaines de virus qui infectent l’épithélium des vertébrés [15]. Les PVs chez les humains sont appelés virus du papillome humain (VPH). Le VPH est la principale cause du cancer du col utérin [16, 17]. Ce cancer est le deuxième cancer le plus fréquent chez les femmes et 80 % des cas survenus principalement dans les pays en développement [18]. Le VPH est un virus composé d’ADN circulaire à double brin d’environ 8 kb qui contient généralement 8 gènes (voir Figure 2). L’un de ces gènes codant la protéine L1 qui forme la capside, est utilisé comme élément de conservation entre les différentes souches du virus VPH, notamment dans les études qui ont mené au développement des vaccins actuels [19].
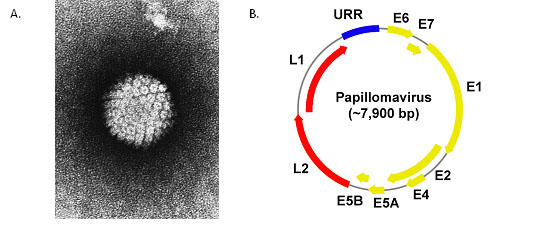
Figure 2 : Visualisation du virus VPH A. capside viral vue par microscopie électronique. B. Schématisation du génome du VPH [17].
Classification des PVs
Les PVs sont très variés. Ils forment une famille qui ne comporte pas de sous-famille. Traditionnellement, les PVs ont été classifiés en types, sous-types et/ou variants [20]. Cette classification n’a pas été initialement reconnue par le comité ICTV qui les classifie officiellement dans la famille Papillomaviridae, composée de 30 genres et de 14 espèces selon le genre (Figure 3). Jusqu’à maintenant, environ 175 types de PVs ont été détecté chez l'humain qui se retrouvent dans les genres alpha, beta, gamma, mu et nu [20-22].
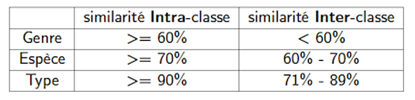
Le critère de classification très largement utilisé pour classifier les PVs est la similarité des séquences d'ADN du gène L1. L1 est le gène le plus conservé dans le génome du PV et donc il a été utilisé pour l’identification de nouveaux types de PVs pendant des années. En 2004, de Villiers et al. ont déclaré une similarité de plus de 60% entre les spécimens d'un même genre, et donc une similarité inter-genre de moins de 60%. La similarité intra-espèce déclarée dans leur étude est de plus de 70%, et donc deux spécimens appartenant à des espèces différentes d'un même genre auront une similarité de 60% à 70%. Ils ont aussi déclaré une similarité intra-type de 90% ou plus, et donc une similarité inter-type de 71% à 89% (Table 1, Figure 4).
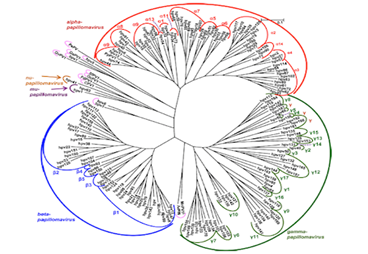
Figure 3: Analyse phylogénétique basée sur les séquences L1 ORF de 170 types de VPH , ainsi que papillomavirus animaux individuels, en utilisant la méthode du maximum de vraisemblance. L'arbre a été construit en utilisant le programme MEGA5.1 [21].
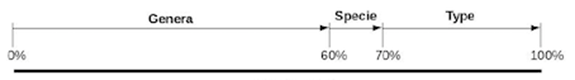
Figure 4. Répartition des classes selon la similarité [6].
Table 1. Caractéristiques de la classification selon la similarité du gène L1 [6].
L'intérêt de refaire la classification
Aujourd'hui avec le développement des nouvelles techniques de laboratoire, le nombre de nouvelles espèces potentielles de papillomavirus à identifier et caractériser a beaucoup augmenté [21]. De plus, avec le nombre de génomes séquencés qui a explosé [6], il faut s’interroger sur la validité de la classification préalablement établie et développer une nouvelle classification qui incluent toute cette nouvelle information, d’où le grand intérêt des travaux présentés lors du séminaire exposés dans ce rapport.
En 2013, Daigle a développé une méthode pour reconstruire un arbre où les feuilles sont les séquences génomiques, et où les nœuds internes de même hauteur correspondent à la hiérarchie des différentes classes (genre, espèce, type). Il a analysé cet arbre en étudiant l'information taxonomique extraite des 560 séquences de VPH obtenues de la base de données GenBank du NCBI. Des programmes informatiques développés ont été utilisés pour vérifier la conformité des classifications par rapport au critère de similarité. Chaque séquence génomique a donc été aligné avec les 559 autres, les alignements multiples ont été réalisés avec l’outil Muscle et « épuré » avec le programme Gblocks. Ensuite, la dissimilarité a été calculée pour chaque paire d’alignement (matrice de dissimilarité triangulaire de 560 x 560).
Les résultats présentés ont été analysés selon trois critères : la distribution de la similarité, la séparabilité et compacité des clusters ainsi que la reproductibilité de la classification.
Distribution de la similarité
En 2010, afin de classifier 189 types de PV, Bernard et al. ont comparé la distribution de la similarité du gène L1 entre les classes intra-espèce (à l’intérieur d’une même espèce), inter-espèce (entre différentes espèces dans un même genre) et inter-genre (entre les genres différents) [15] (Figure 5). En 2013, Daigle a étudié ce critère en utilisant 560 séquences de VPH sur le gène L1 (Figure 6), ainsi que sur le génome complet (Figure 7) [6, 22].
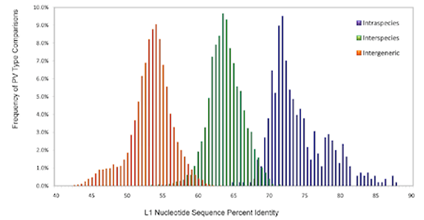
Figure 5 Comparaisons spécifiques inter-genres, inter-espèces et intra-espèces des séquences de nucléotides de la région L1 selon la matrice d’alignement multiple des séquences (189 PVs) [15].
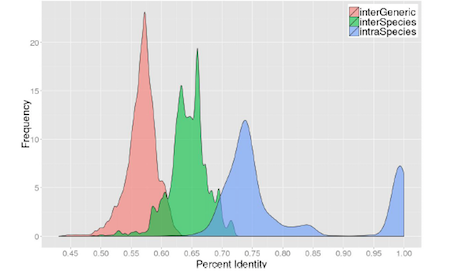
Figure. 6: Comparaisons spécifiques inter-genres, inter-espèces et intra-espèces des séquences de nucléotides de la région L1 selon la matrice d'alignement multiple des séquences (560 VPHs) [6].
En comparant les graphes, celui de la distribution de la similarité du gène L1 des VPHs (Figure 6) est plutôt similaire à celle du graphe du gène L1 des PVs (Figure 5). Dans les deux cas, il y a un chevauchement des courbes signifiant une zone dans lesquelles la séparation des différents groupes n’est pas optimale. De plus, dans la comparaison intra-espèce (aire bleue dans la Figure 6), on peut noter un sous-groupe de données qui a un pourcentage d’identité de 100%. Ce phénomène est relié à l’utilisation d’un plus grand pool de séquences pour générer les résultats du graphe de la figure 6 et dans ce pool il y a un certains nombres de séquences qui sont identifiques d’où l’apparition de la courbe à l’extrême droite du graphique. La distribution produite pour le gène L1 (Figure 6) est semblable de celle pour le génome entier (Figure 7). Toutefois, il y a des différences, qui ne devraient pas se produire selon de Villiers et al. (2003), qui méritent d’être soulignées. C’est-à-dire que l’utilisation du génome complet permet de diminuer les chevauchements entre les 3 classes (genre, espèce et type), ce qui est illustré par une zone de chevauchement plus petites entre les différentes courbes. De plus, le graphe de la figure 7 démontre des groupes plus compacts de valeurs ce qui dénote une meilleure délimitation des classes avec cette méthode.
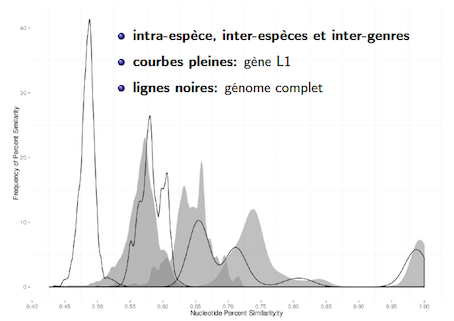
Figure 7 : Comparaisons spécifiques inter-genres, inter-espèces et intra-espèces des séquences de nucléotides de la région L1 vs du génome complet selon la matrice d'alignement multiple des séquences (560 VPHs) [6].
Séparabilité et compacité des classes
La classification du VPH est un regroupement (clustering) hiérarchique où les classes sont les noeuds internes du dendrogramme. Dans un regroupement, il est important que les éléments à l'intérieur d'un sous-groupe soient similaires (compacité), et que ceux des sous-groupes externes soient distincts (séparation). À l'aide d'une mise à l'échelle, il est possible de visualiser la distance entre les séquences pour avoir une idée de la compacité et de la séparation des clusters. Les tendances de regroupements sont aussi essentielles pour classifier les nouveaux échantillons biologiques [23] (Figure 8).
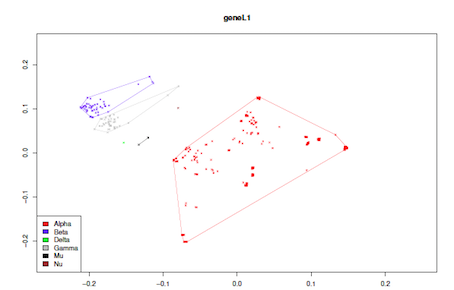
Figure 8: Mise l’échelle multidimensionnelle des dissimilarités, cernées par l’enveloppe convexe de chaque genre, ne montre pas de chevauchement entre les genres, mais les genres gamma et beta sont très rapprochés [6].
Il existe plusieurs méthodes de visualisation des clusters. Le graphe présenté dans la figure 8 a été généré via l’utilisation du langage R, en utilisant un script « maison » qui emploie les packages cmdscale () pour la projection, plot () pour générer le graphe et chull () pour l’enveloppe. L’échelle multidimensionnelle est nécessaire pour visualiser l’ensemble des clusters simultanément. En effet, dans une échelle unidimensionnelle, les clusters estimés ne sont pas nécessairement tous visibles, il est donc difficile d’évaluer la séparation des groupes (voir Figure 9 A).
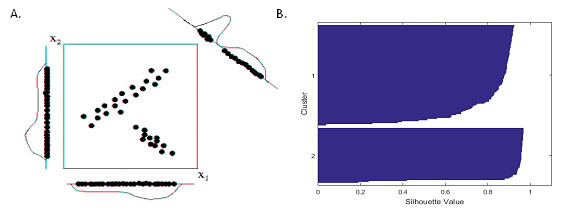
Figure 9: Visualisation de la séparation et compacité des clusters.A. graphe représentant les clusters sur une échelle unidimensionnelle (axe x1 et x2) vs multidimensionnelle (graphe x1/x2) [24]. B. Un graphe Silouhette qui démontre l’importance de la taille des clusters (plus un cluster contient des points plus il est imposant) et la séparation entre les clusters (espaces entre les clusters) [25].
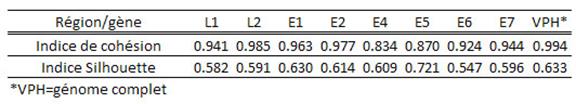
Plusieurs méthodes existent pour analyser et schématiser la séparation de clusters (voir Figures 8 et 9 B). Il est possible de le faire à partir d’indice qui mesure le degré de séparation et/ou compacité et/ou cohésion. Dans le projet présenté par Bruno Daigle, l'indice de cohésion a été créée afin de mesurer le degré de compacité et de séparation et d'évaluer la robustesse de la classification des VPHs [6]. Cet indice est dérivé de l'indice Silhouette [26]. Alors que l'indice Silhouette fait la comparaison en base de la distance moyenne entre les éléments, mais l'indice de cohésion utilise une valeur booléenne, donc il est plus facile à interpréter et moins biaisé par rapport à la distance. Il mesure le degré de compacité et de séparation selon que la similarité minimale intra-classes est ou non inférieure à la similarité maximale interclasses, pour chaque paire de séquences et pour chaque classe. L'indice de cohésion est maximum (0.994) lorsque la classification est effectuée avec le génome complet [6].
La conclusion du projet de M. Daigle concernant la séparation des clusters est que l'augmentation du nombre des génomes séquencés a un effet bénéfique sur la compacité et la séparation des sous-groupes, mais peu sur les arbres phylogéniques, surtout avec utilisation des séquences plus longues comme le génome complet [6].
Reproductibilité
La reproductibilité est un paramètre important à considérer lors des analyses bio-informatiques. La quantité de données phylogénétiques générées a explosé dans les dernières années notamment à cause de l’amélioration de l’accessibilité aux technologies de séquençage. La reproductibilité permet entre autres d’avoir une standardisation du protocole. Une des approches possibles est de développer des flux de travaux dans des environnements « open-source » convivial et accessible à tous. Différents systèmes de gestion des flux de travaux peuvent être utilisés afin de générer des pipelines qui facilitent la reproductibilité des résultats, tels que : Kepler, Gene Prof, Taverna, Armadillo, Galaxy, etc. [27] Certains systèmes sont mêmes spécialisés dans la reproductibilité des analyses phylogénétiques, comme par exemple Phylogenerator, ReproPhylo ou Osiris [28, 29].
La reproductibilité permet également de valider une nouvelle approche développée, en utilisant un jeu de données similaires provenant d’une autre étude et en tentant de reproduire les résultats obtenus précédemment. Les travaux présentés lors de la conférence étaient directement en lien avec les travaux présentés par l’équipe de De Villiers [20]. Bruno Daigle a donc choisi de tester la reproductibilité en deux étapes. La première étant de vérifier l’impact de l’utilisation d’un plus grand nombre de génomes. La classification phylogénétique à l’aide du gène L1, telle que décrite dans l’étude de 2004 a donc été refaite, mais avec 560 séquences au lieu des 190 séquences utilisées originalement. Les arbres ont été générés par la technique de maximum de vraisemblance avec le logiciel phyML, avec le modèle de substitution HKY85 pour construire les matrices de distances et une analyse de bootstrap avec 128 itérations. Dans ce cas, la classification a été reproduite. Toutefois, les scores de bootstrap diminuent quand le nombre de séquences utilisées augmente. Ce phénomène pourrait être dû au fait quand le nombre total de séquences augmente, il y a plus de séquences qui se ressemblent, alors les possibilités de décisions de l’algorithme d’inférence de l’arbre sont plus vastes car il y a plusieurs possibilités équivalentes. Dans la seconde étape, la classification générée par l’utilisation des séquences d’un seul gène L1 a été comparé avec la classification généré à l’aide des génomes complets, afin d’évaluer si les classifications étaient semblables ou non, réussit-on à reproduire la même classification en utilisant un seul gène ou plusieurs gènes? Pour évaluer ce paramètre différentes méthodes pourraient être envisagées, notamment de calculer la distance de Robinson-Foulds (RF) entre les deux arbres générés. Puisque la distance RF calcule le nombre minimal d’opérations permettant d’obtenir l’arbre B à partir de l’arbre A, plus son score est faible, plus les arbres sont similaires) [30]. La distance RF a été utilisée pour comparer les deux arbres phylogéniques du gène L1 et du génome complet avec l'arbre taxonomique. Suite à cette analyse, il a été conclu que les deux arbres sont identiques à l'arbre taxonomique et donc, à la classification actuelle. La différence est entre le degré de confiance qui est faible pour le gène L1 et plus élevé pour le gène complet, ce qui suggère d'utiliser le génome complet pour une inférence phylogénique optimale [6]. Les scores de bootstrap obtenus sont plus élevés quand le génome entier est utilisé versus la séquence d’un gène, ce phénomène est attendu puisque la séquence génomique étant plus longue, le nombre de variations augmentent par rapport à la séquence génique et permet ainsi à l’algorithme d’inférence d’avoir une solution plus nette.
Tel que décrit dans ce rapport, les résultats présentés lors du séminaire ont été analysés selon trois critères soit : la distribution de la similarité, la séparabilité et compacité des clusters ainsi que la reproductibilité de la classification. L’utilisation de génomes complets pour établir la classification des virus VPHs a permis de diminuer le chevauchement des frontières et donc de mieux délimiter les classes (genre, espèce et type). Le développement de la mesure de l’indice de cohésion soutient également l’idée selon laquelle la classification à l’aide du génome complet fournit une meilleure classification via une séparabilité et compacité optimale (indice de cohésion maximal par rapport à l’utilisation des gènes de façon individuelle). La méthode utilisée a permis de reproduire la classification qui avait été préalablement établie et de l’améliorer en utilisant le génome entier et un plus grand nombre de séquences. L’ajout de plus de séquences ne permet pas d’améliorer la classification faite à partir des gènes de façon individuelle. L’augmentation du nombre total de séquences augmente aussi le nombre de possibilités d’inférence, puisque le nombre de séquences très similaires augmente aussi. La solution à ce problème est d’utiliser une séquence plus longue (une combinaison de gènes ou encore le génome complet tel que brillamment illustré dans ce séminaire) afin de minimiser ce phénomène.
La classification phylogénique des papillomavirus humains a permis de remettre en questions l’ancienne classification phénotypique basée notamment sur les sites d’infections, le partitionnement VPHs muqueux vs VPHs cutanés n’est pas soutenu par la classification phylogénique. Depuis 2004, la classification ICTV des virus papillomes humains est basée sur les classes définies par la classification phylogénique [21].
Les perspectives concernant ce projet sont très intéressantes et concernent notamment une automatisation de la classification. Tel que discuté dans la section reproductibilité, l’utilisation de plateformes disponible pour tous (Armadillo, Taverna) afin de générer un flux de travail efficace permettrait d’uniformiser la classification des virus VPHs et de simplifier l’ajout de nouveau taxon. De plus une optimisation pourrait être faite dans la genèse des arbres phylogénomiques en utilisant des programmes de maximum de vraisemblance plus optimaux pour les grandes quantités de données d’entrée tels que RAxML ou Fast Tree [31, 32]. Dans l’analyse présentée par B. Daigle, le niveau de séparation est illustré par l’indice de cohésion. La formule utilisée par B. Daigle démontre ici que la classification optimale est obtenue via l’utilisation des génomes complets et non des séquences de gènes individuels. Toutefois, il existe diverses façons de valider le clustering de données, par exemple les méthodes Silhouettes, SSE (sum of squares error) et calculs d’entropie [26]. Si la méthode Silhouette est utilisée sur les données présentées, le résultat maximal est obtenu avec l’utilisation du génome complet tout comme avec l’indice de cohésion (voir table 2). Ce qui confirme que le génome entier permet d’avoir une classification plus précise que les gènes individuels.
Table 2 : Comparaison des indices pour valider le clustering
Le pourcentage de similarité a été utilisé comme critère de classification toutefois, la ségrégation des classes pourrait être améliorée en combinant d’autres critères, tels que des critères de classification phénotypique. Les paramètres qui pourraient être utilisés regroupent l’indice de pathogénicité/oncogénicité (variant sur une échelle de valeurs entre aucun jusqu’à très haut risque), sites d’infections (peau, muqueuse), attributs des protéines virales (poids moléculaire, poids isoélectrique, structures tertiaires / quaternaires, domaines d’interactions, etc.).
En terminant, il est important de mentionner que certains outils bio-informatiques, tels que PaVE qui consiste en une base de données et un regroupement d’applications web utiles dédiés à l’étude des papillomavirus sont très utiles pour l’avancement des connaissances sur le sujet [33].
3. http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?name=Viruses.
13. http://evolution.berkeley.edu/evolibrary/news/060101_batsars.
14. Sharp, P.M. and B.H. Hahn, AIDS: prehistory of HIV-1. Nature, 2008. 455 (7213): p. 605-6.
25. http://www.mathworks.com/help/releases/R2015a, Cluster analysis. 2015. R2015a.
28. http://willpearse.github.io/phyloGenerator/.