Rapport
BIF7002
Analyse de
recombinaisons et de transferts horizontaux de gènes chez SARS-CoV-2
Stéphane Samson et Vladimir Makarenkov
Université du Québec à Montréal
Présenté par:
Omar Al Rifai
21-2-2023
Table of Contents
4- La fonctionnalités de recherche de site
I-
Introduction
Le transfert horizontal des gènes (HGT) est le
transfert de matériel génétique entre deux espèces différentes. C'est un
processus important pour acquérir du nouveau matériel génétique et pour évoluer
vers de nouveaux aspects génétiques. Ce processus se produit à travers trois
mécanismes différents: la transformation, la conjugaison et la transduction [1].
La transformation est le processus par lequel les bactéries (cellules
réceptrices) prennent de l'ADN libre neuf, la conjugaison est le
processus par lequel les bactéries transfèrent leur matériel génétique à
d'autres cellules, et la transduction est le processus par lequel un virus de
bactériophage transfère du matériel génétique entre des cellules. Le processus
de HGT a été décrit pour la première fois en 1951 dans le bacille de Klebs-Löffler, où le gène toxique responsable de sa pathogénicité
peut être transféré des bactéries pathogènes à des bactéries non pathogènes [2].
Il peut impliquer le transfert de matériel génétique entre différentes espèces,
telles que les bactéries, les virus et les cellules eucaryotes. Par exemple, l’HGT
est un processus par lequel certaines bactéries peuvent acquérir une résistance
aux antibiotiques et développer de nouvelles souches, comme la souche CC398 de
Staphylococcus aureus. Des études récentes ont montré l'existence de transfert
de gènes entre des cellules virales et des cellules eucaryotes, tandis que le
transfert de gènes d'eucaryotes à des virus se produit deux fois plus
fréquemment. L’HGT implique des virus à ADN double brin, en particulier les
NCLDV (virus à ADN nucléocytoplasmique de grande taille), ainsi que les herpèsvirus, les rétrovirus et les bactériophages [3].
Le syndrome respiratoire aigu sévère coronavirus 2
(SARS-CoV-2) est une maladie infectieuse respiratoire apparue à la fin de
l'année 2019. Sa transmissibilité très élevée a entraîné sa propagation
pandémique dans le monde entier. La pandémie a été appelée pandémie de
COVID-19. Ce dernier n'était pas le seul coronavirus du XXIe siècle, en 2002 et
2012, SARS-CoV et MERS-CoV sont apparus chez
l'humaine respectivement. SARS-CoV-2 appartient à la famille des
bêta-coronavirus, il partage 96% de similarité avec le coronavirus de la
chauve-souris RatG23 et 90% de similarité avec les coronavirus de pangolin [4].
SARS-CoV-2 partage six régions avec les bêta-coronavirus, l'ORF1a, l'ORF1b, la
protéine Spike (S), l'enveloppe (E), la membrane (M) et le nucléocapside (N),
ainsi que 7 ORF codant pour des protéines accessoires (Figure 1) [5, 6].
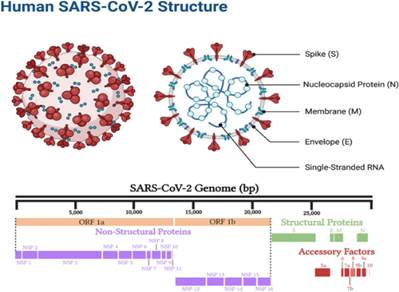
Figure 1- Les composants qui constituent la structure du virus
comprennent la protéine de spike, l'enveloppe, la membrane, ainsi que des
composants internes tels que l'ARN monocaténaire viral et les protéines de
nucléocapside.
Une caractéristique spécifique du SARS-CoV-2 est la
présence d'un motif de base (PRRA), qui est un site de clivage de la furine de la proprotéine
convertase. Ce motif relie les sous-unités spike S1 et S2 [5].
Un motif très similaire ne contenant pas (PAA), les résidus d'acides aminés de
base (RR) ont été trouvé dans le coronavirus RmYN02 dérivé de la chauve-souris [7].
Ces données suggèrent que le SARS-CoV-2 est très probablement apparu
naturellement. Trois hypothèses différentes ont été proposées pour expliquer
l'évolution du SARS-Cov2. La première est l'évolution parallèle où les
différences génétiques entre les souches de virus pourraient être acquises par
des mutations naturelles survenues lors des passages et de l'évolution du
virus. La deuxième hypothèse suggère la divergence de GD pangolin Cov et de SARS Cov2 à partir d'une ascendance similaire. La
troisième hypothèse propose une possibilité de transfert génétique et de
recombinaison intergénique dans le génome du virus SARS Cov
chez l'espèce hôte. Cette recombinaison nécessite une espèce donneuse et une
autre accepteuse. Elle conduit à un nouveau virus avec une structure génique en
mosaïque où chaque fragment de gène est dérivé d'une évolution différente du
virus. Pour répondre à la question de savoir si le transfert horizontal de
gènes contribue au mécanisme par lequel le SARS-CoV-2 se développe et diverge,
Makarenkov et al. ont développé l'algorithme SimPlot pour enquêter sur les
origines du virus en détectant et en évaluant les événements de transfert
horizontal de gènes et de recombinaison en utilisant la séquence de différentes
souches de virus SARS-CoV.
II-
Méthode
SimPlot++ est une
application open source gratuite qui est implémentée en Python et qui offre une
gamme d'outils pour l'analyse de séquences. Elle est utilisée pour visualiser
et analyser les relations entre les séquences de nucléotides ou d'acides
aminés. Elle peut afficher des arbres phylogénétiques, des alignements de
séquences et des diagrammes de similarité, qui montrent le degré de similarité
entre des paires de séquences à chaque position sur leur longueur. De plus, Elle peut identifier les événements de recombinaison intergéniques et intragéniques à l'aide des tests Phi, χ2, NSS et
Proportion. SimPlot++ prend également en charge le multitraitement et fournit
des diagnostics en calculant des distances. Les autres fonctionnalités incluent
la création et l'enregistrement de séquences consensus, l'exécution d'une
analyse BootScan, l'identification de sites
informatifs et l'analyse de réseaux de similarité de séquences.
1-
La création de groupe
La page de création de groupe dans Simplot ++ accepte plusieurs fichiers de séquence dans différents formats tels que Fasta, Nexus, Pir, Phylip, Stochholm ou Clustal. Ces séquences sont organisées manuellement dans le groupe, SimPlot ++ générera une séquence consensus pour chaque groupe. Cette séquence consensus dépend du seuil déterminé par l'utilisateur. L'analyse du consensus est essentielle pour l'analyse Simplot supplémentaire telle que le bootscan et l'analyse de similarité de réseau.
Figure 2- Démonstration de la création de groupe
Grâce au navigateur de fichiers, les utilisateurs peuvent télécharger leur séquence d'ADN ou d'acides aminés (AA) dans le format accepté (Figure 2). L'ID de séquence apparaîtra dans la section de séquence non groupée où ils peuvent être ajoutés à un groupe spécifique. Lorsque les groupes sont prêts, il est recommandé de les enregistrer au format Nexus pour éviter de recréer le groupe à nouveau. La création de groupe est requise avant de commencer l'analyse à l'aide des fonctionnalités de SimPlot ++. 2-
L'analyse Simplot
L'analyse Simplot génère une matrice de distance de l'alignement de séquences multiples (Figure 3). Il est essentiel de déterminer la fenêtre de distance d'analyse et la taille du pas. Cette matrice est visualisée à travers un graphique de similarité qui aidera à détecter d'éventuels événements de recombinaison. Pour utiliser les outils d'analyse Simplot, il est important de choisir un paramètre qui correspond aux objectifs de l'analyse. Cela comprend la détermination de la longueur de la fenêtre, la taille du pas, la bande GAP, le modèle de distance et précise l'utilisation ou non du multitraitement qui permet l'exécution simultanée d'analyses multiples sur un grand ensemble de données.
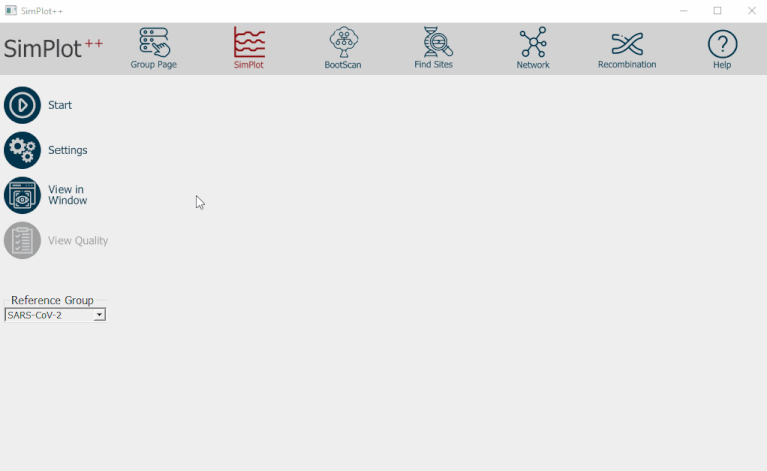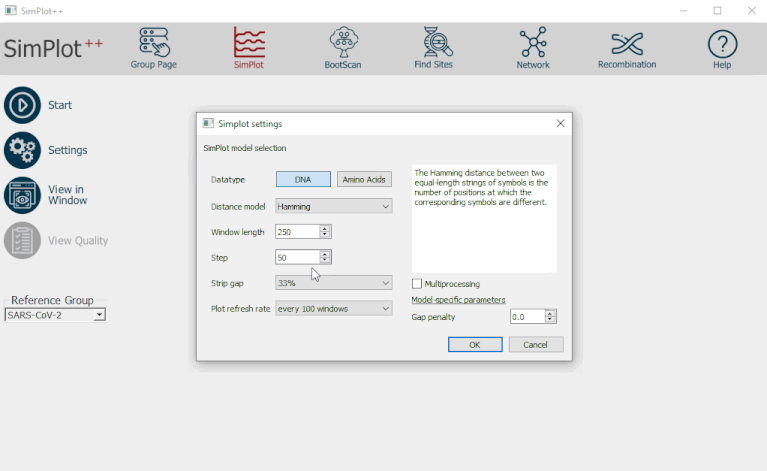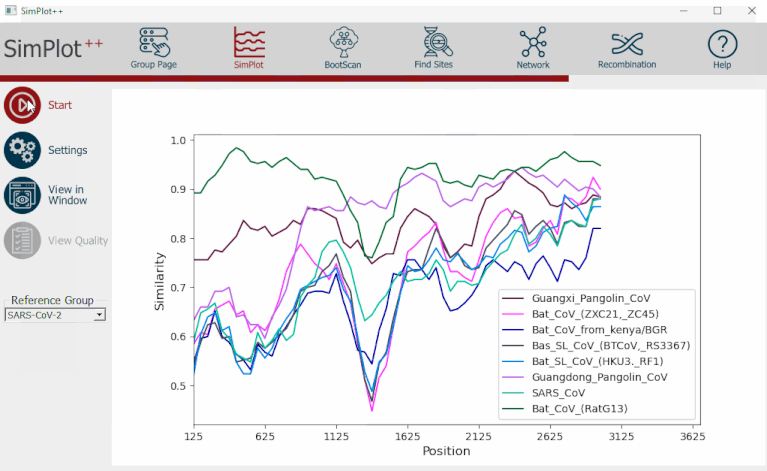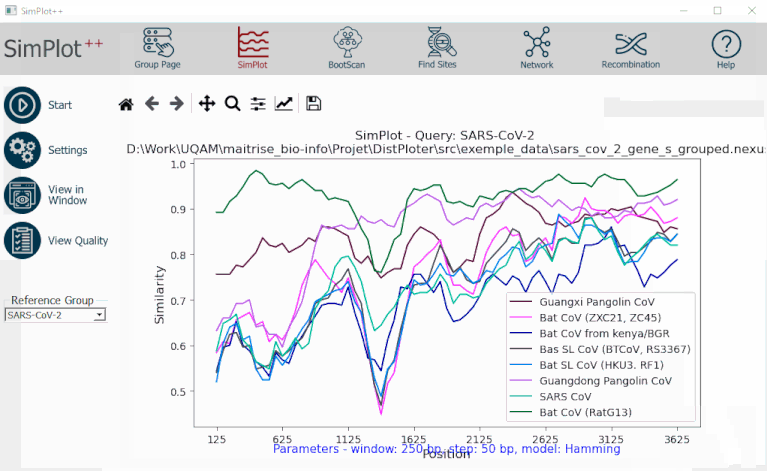
Figure 3- Démonstration de l’analyse SimPlot
SimPlot ++ a une fonction de contrôle qualité supplémentaire pour visualiser les données liées aux écarts. De plus, Simplot ++ a la fonction d'analyse du réseau de similarité de séquence, une représentation interactive d'une analyse SimPlot, où chaque groupe est représenté comme un nœud dans un réseau, connecté par un bord basé sur la similarité globale ou locale (Figure 4). En ajustant le seuil minimum de similarité, les relations entre les groupes peuvent être analysées. Les tableaux de données montrent les régions de similarité les plus importantes et les résultats du test de proportion. Le graphique peut être enregistré sous forme de fichier HTML et la visualisation peut être enregistrée sous forme de fichiers .png ou .svg à partir de la boîte à outils du fichier HTML.
Figure 4- Démonstration de l'analyse du réseau de similarité de séquence
3-
La fonctionnalité Bootscan
La fonctionnalité Bootscan est un pipeline d'analyse de fenêtres glissantes composé de quatre étapes principales (Figure 5). Tout d'abord, les sous-séquences sont isolées des groupes consensus et amorcées N fois. Deuxièmement, une matrice de distance est générée pour chacun des N sous-MSA bootstrapés. Troisièmement, pour chaque matrice de distance, un arbre phylogénétique est généré soit en utilisant Neighbor-Joining ou UPGMA. Les signaux phylogénétiques conflictuels sont quantifiés en pourcentage de chaque séquence au voisin le plus proche de la séquence de référence dans l'arbre. Bootscan a un paramètre similaire à l'analyse Simplot, tel que le paramètre des fonctionnalités d'analyse et la visualisation des tracés, en plus du modèle d'arbre et du format de sortie multiple.

Figure 5- Démonstration de l'analyse du Bootscan
4-
Les fonctionnalités de recherche du site
Les fonctionnalités de recherche du site localisent les sites de recombinaison en fournissant deux séquences supplémentaires provenant de chacun des sites parentaux et une séquence provenant d'un groupe différent (Figure 6). Enfin, la fonction de recombinaison permet de détecter les événements de recombinaison et de déterminer leur signifiance sur la base des tests Phi, Phi-profile, Max χ2 et NSS disponibles dans le package PhiPack développé par Trevor Bruen et al. (Figure 7) [8].
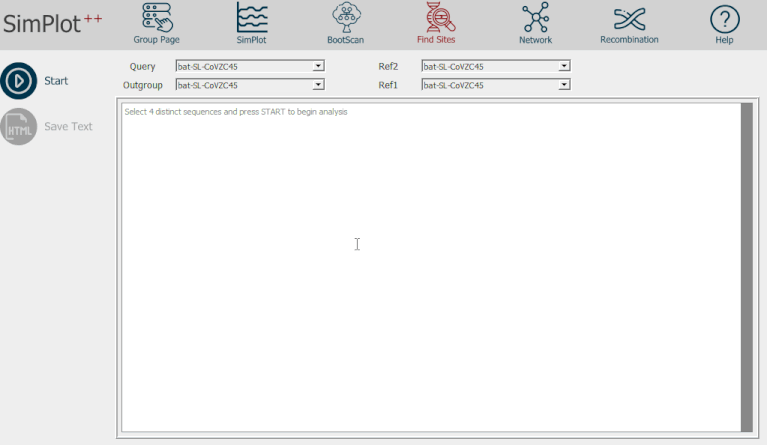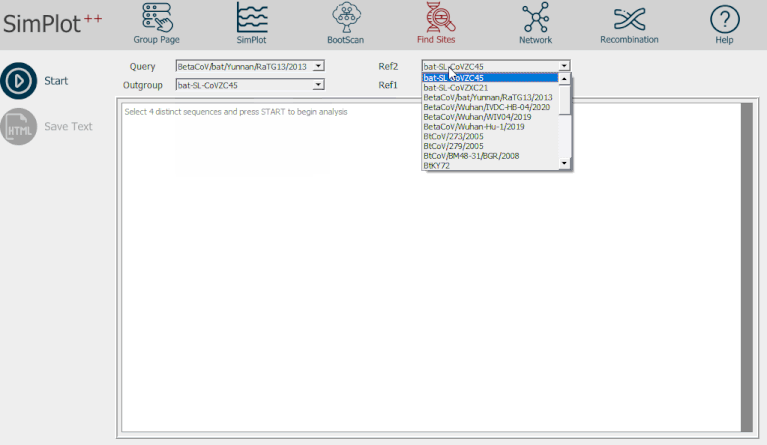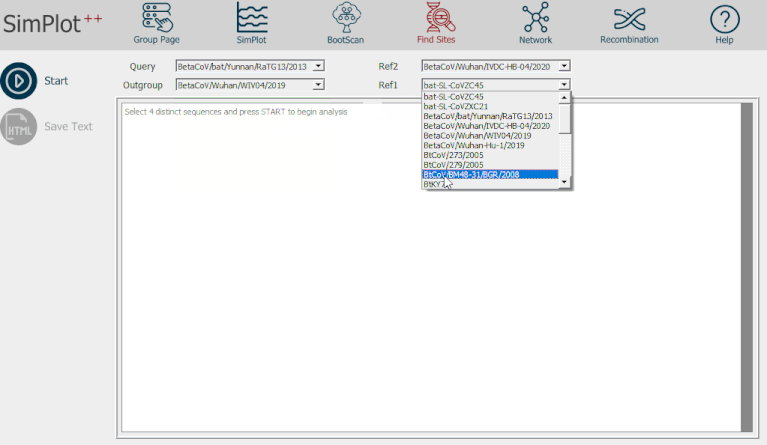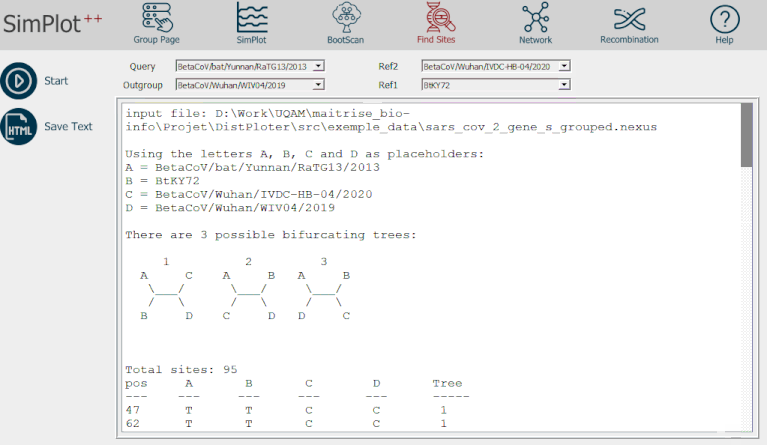
Figure 6- Démonstration de recherche de site
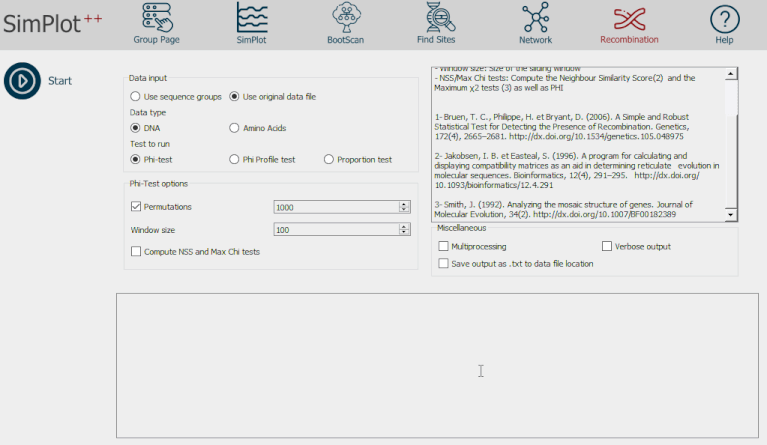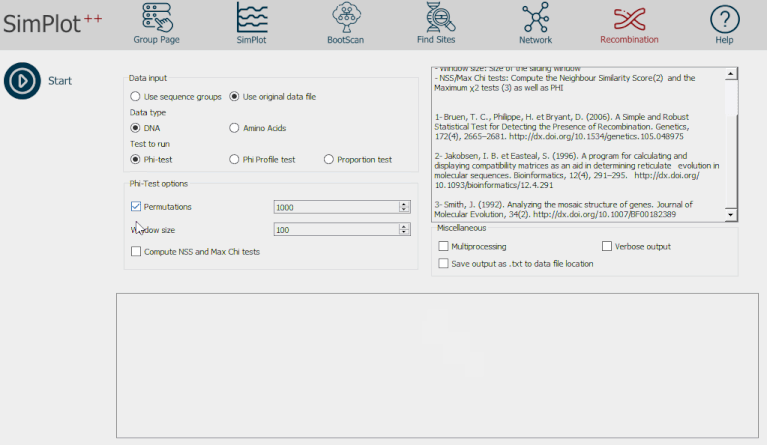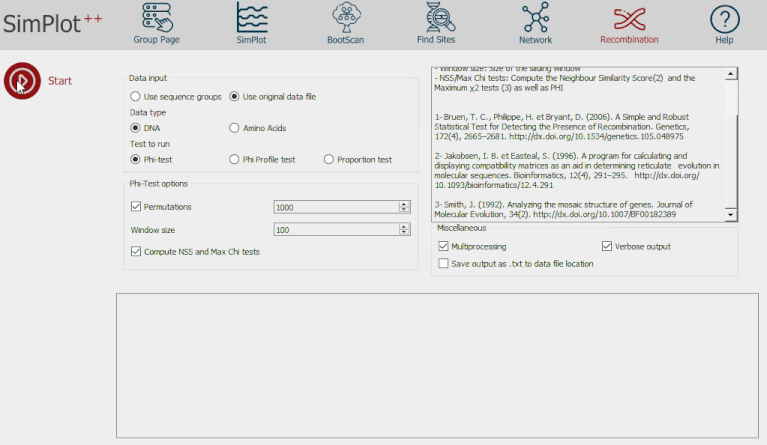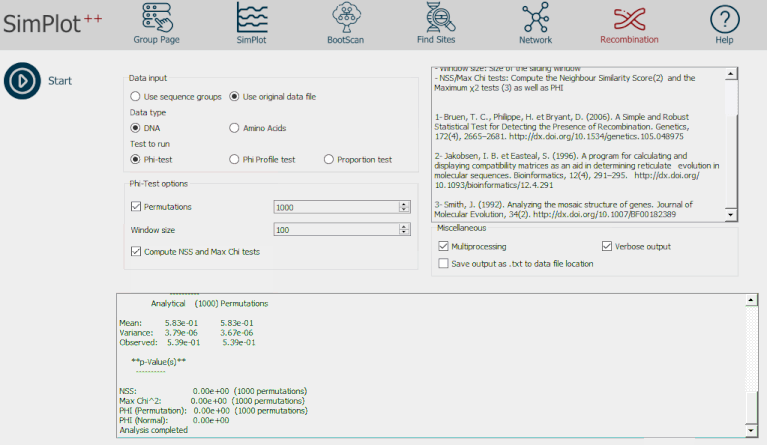
Figure 7- Démonstration de recombination
III-
Results:
L'analyse SimPlot de 25 génomes CoV a révélé que les génomes Wuhan SARS-CoV-2 et RaTG13 partagent 96,14 % de l'identité du génome entier, tandis que les génomes Wuhan SARS-CoV-2 et GD Pangolin sont identiques à 90,34 % (Figure 8) [9]. Cela indique que les chauves-souris sont probablement le réservoir d'origine du SARS-CoV-2, étant donné la ressemblance étroite entre le SARS-CoV-2 et le RaTG13, comme ce fut le cas lors des précédentes épidémies de CoV.
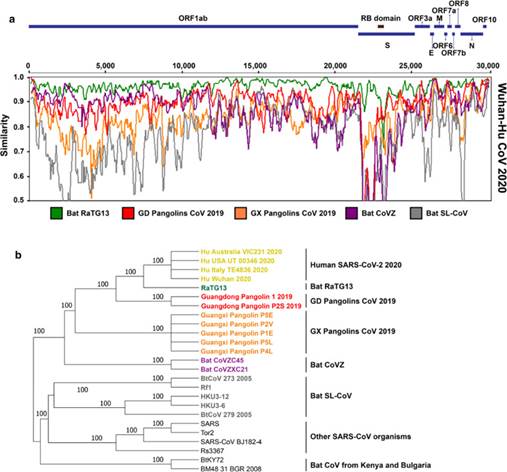
Figure 8- Similitude du génome et analyse phylogénétique du SRAS-CoV-2 et des virus apparentés : (a) une analyse de l'évolution des modèles de similarité de séquence par SimPlot entre : le génome de référence Wuhan SARS-CoV-2 2020 avec le génome RatTG13 CoV (vert), GD Pangolin CoV (rouge), GX Pangolin CoV (orange), Bat CoVZ (violet) et Bat SL-VoC (gris). (b) Analyse phylogénie du génome de 25 organismes SARS-CoV et au SARS-CoV-2. Les groupes d'espèces sont indiqués à droite. Les scores bootstrap sont indiqués sur les nombres sur le branches internes, ceux qui sont inférieur à 60% ont été effondrées.
Une analyse gène par gène a été effectuée en comparant les séquences génétiques du Wuhan SARS-CoV-2 avec les séquences des groupes RaTG13, GD Pangolin CoV et Bat CoVZ. L'analyse a révélé que certaines régions du domaine RB, ORF3a, M ORF7a et N du SARS-CoV-2 étaient plus similaires au GD Pangolin CoV qu'au RaTG13, suggérant des événements de recombinaison entre les deux (Figure 9). De plus, dans certaines régions géniques continues d'ORF1ab, ORF3a, M et N, le SARS-CoV-2 s'est trouvé plus similaire aux virus bat CoV ZC45 et ZXC21 qu'auxGD Pangolin CoV et parfois même au RaTG13 [9].
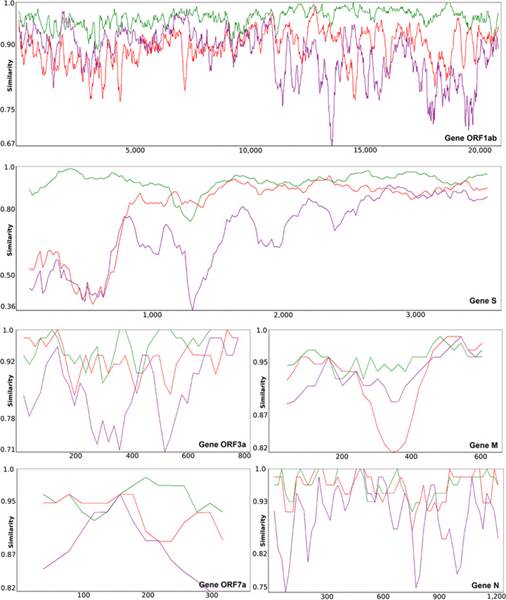
Figure 9- Analyse de similarité SimPlot pour comparer les séquences de gènes ORF1ab, S, ORF3a, M, ORF7a et N du génome de référence Wuhan SARS-CoV-2 2020 avec celles du génome RatTG13 (vert), GD Pangolin CoV (rouge) et Bat CoVZ (violet).
Makarenkov et al, ont effectué une analyse détaillée pour détecter les similitudes du gène Wuhan SARS-Cov-2 avec les séquences du groupe RaTG13, du groupe GD Pangolin CoV et du groupe Bat CoVZ. Cette analyse a révélé queleSRAS-CoV-2 présente des similitudes plus élevées avec le virus GD Pangolin que les régions RaTg13 dans le domaine RB, ORF3a, M, ORF7a et N suggérant qu'un événement de recombinaison multiple s'est produit entre le virus GD Pangolin et le virus RatG13 CoC. Pour valider davantage ces données, Makarenkov et al. ont effectué un test d'analyse de recombinaison gène par gène Φ-test pour évaluer l'importance de la recombinaison entre les espèces orthologues de SARS-CoV-2, RaTG13, GD Pangolin 1 Cov, GD Pangolin P2S CoV, bat CoV Virus ZC45 et bat CoV ZXC21 (Tableau 1). Ce test révèle une recombinaison significative au niveau des ORF1ab, S, ORF3a, ORF7a, ORF8, N et de l'ensemble du génome [9].
|
Region |
Φ-test result |
Recombination detected (yes/no) |
Window size |
|
Gene ORF1ab |
2.23 × 10–3 |
Yes |
150 |
|
RB domain |
Too short |
– |
|
|
Gene S |
8.60 × 10–3 |
Yes |
200 |
|
Gene ORF3a |
1.13 × 10–5 |
Yes |
100 |
|
Gene E |
0.56 |
No |
200 |
|
Gene M |
0.226 |
No |
100 |
|
Gene ORF6 |
Too short |
– |
|
|
Gene ORF7a |
0.00155 |
Yes |
200 |
|
Gene ORF7b |
Too short |
– |
|
|
Gene ORF8 |
0.0453 |
Yes |
200 |
|
Gene N |
0.024 |
Yes |
50 |
|
Gene ORF10 |
Too short |
– |
|
|
Whole genomes |
0.0156 |
Yes |
200 |
Tableau 1- Φ-test pour les évènements de recombinaison
Le génome de SARS-CoV-2 est composé de 2 gènes codant pour 2 protéines non structurelles ORF1a et ORF1 b, 4 gènes codant pour 4 protéines structurales (S, E, M, N) et 5 gènes codant pour 5 protéines accessoires (ORF3, ORF6, ORF7a, ORF7b, ORF8, ORF10). ORF1ab est traduit de ORF1a (11826–13425 nt) et ORF1b (7983–8157 nt) [10, 11] . Le gène S code pour la protéine de Spike qui a une structure en forme de spike. La protéine de pointe est une glycoprotéine de type 1 essentielle pour la liaison au récepteur, la fusion et l'entrée du virus. Le gène E est commun à tous les CoV, le gène M est important pour l'assemblage de nouvelles particules virales. La protéine de nucléocapside est codée par le gène N et c'est un composant structurel essentiel du SRAS-CoV. Il est responsable de l'encapsulation du génome viral. ORF3a, ORF6, ORF7a et 7b, ORF8 et ORF10 sont des protéines accessoires qui ne se trouvent pas dans tous les SARS-CoV. Ils jouent un rôle dans la régulation des gènes, dans l'apoptose et dans l'interaction avec MHC1. Le tableau suivant résume les événements de recombinaison identifiés par Makarenkov et al à travers les différents gènes SARS-Cov2 parmi les différentes espèces (Tableau 2) [9].