TP1 - Julie Cardin et Benoit Fiset
- Hiver 2015
Next Generation Sequencing (NGS) amène de nouvelles perspectives scientifiques
◦ Loi de Moore
◦ Tableau: Quantité de matériel biologique requise, coûts, temps, espace données
◦ Général
◦ Comparaison des méthodes Electrophorèse Capillaire Sanger versus NGS
◦ Extraire l'ADN
◦ Préparation d'ADN
◦ Générer les regroupements
◦ Séquencer en Next Generation
◦ Séquençage avec Référence
◦ Séquençage De Novo
◦ Résolution ou Coverage
◦ Exemples d'applications, Génome entier, Petits génomes
◦ Illumina
◦ Méthode Sanger
◦ Génome Québec
◦ Plate forme séquençage IRCM
◦ Méthylation du génome
◦ Représentation Réduite et Séquençage Bisulfité (RRSB)
◦ Étude Éipgénétique: Projet Dépendance à la cocaïne
1. Introduction
Le 3 Février 2015, nous assistons à la conférence d'Alpha Boubacar Diallo, Ph.D. Chercheur Associé à Institut Universitaire en Santé Mentale Douglas. Dr. Diallo intitule sa conférence « Analyse de données de séquençage deuxième génération ».
L'introduction de Dr. Diallo débute avec une citation d'un document de la compagnie Illumina, fabricant de systèmes de séquençage « Next Generation Sequencing » (NGS).
“Next Generation Sequencing technology
have seen a major transformation in the
way scientists extract genetic information from biological systems,
revealing limitless insight about the genome,
transcriptome, and epigenome of
any species. This ability has catalyzed a number of important breakthroughs, advancing scientific fields from human disease research to agriculture and evolutionary
science.
NGS data output has
increased at a rate that outpaces Moore’s
law, more than doubling each year since it was invented. In 2007, a single sequencing run could produce
a maximum of around one gigabase (Gb) of
data. By 2011, that rate has
nearly reached a terabase (Tb) of data
in a single sequencing run—nearly a 1000×
increase in four years.
With
the ability to rapidly generate large volumes of sequencing data, NGS enables researchers
to move quickly from an idea to full
data sets in a matter of hours or days. Researchers can now sequence more
than five human genomes in a single run,
producing data in roughly one week,
for a reagent cost of less than $5,000
per genome. By comparison, the first human genome required roughly 10 years to sequence using CE
technology and an additional three years to finish the analysis. The completed
project was published in 2003, just a few years before NGS was invented, and
came with a price tag nearing 3 billion
USD. ”
http://www.illumina.com/documents/products/Illumina_Sequencing_Introduction.pdf
Cette citation jette les bases pour notre
travail de rapport de cette conférence. Nous pouvons décrire notre approche
pour notre travail par « D'où viennent ces données et comment nous y
sommes-nous parvenus.»
Un peu d'histoire et de terminologie.
En 1965, Gordon E. Moore, co-fondateur d'Intel Corporation, décrit dans un document une observation sur l'histoire du matériel informatique. Il décrit une tendance et formule sa déclaration actuelle en 1975. La «Moore's Law» est née.
Simplement, «Moore's Law» est l'observation, sur l'histoire du matériel informatique, que le nombre de transistors dans un circuit intégré double environ tous les deux ans. Cela veut aussi dire que la puissance de calcul des ordinateurs augmente aussi.
|
|
Cela est particulièrement pertinent,
pour le domaine de la NGS, car cette puissance de calcul est directement liée
aux besoins de NGS pour recueillir, analyser et gérer tous ces données de
séquençage. |
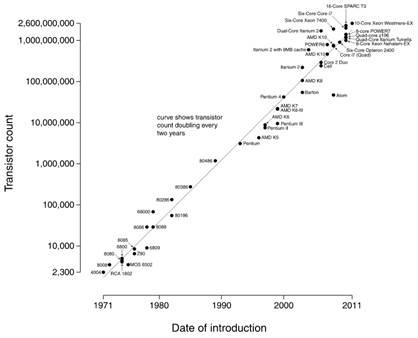
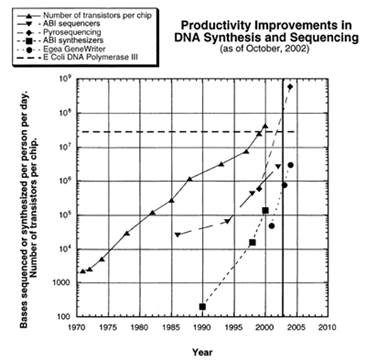
En Septembre 2003, Robert Carlson, chercheur à l'Université de Washington, publie un document intitulé «The Pace and Proliferation of Biological Technologies» (http://www.kurzweilai.net/the-pace-and-proliferation-of-biological-technologies-2).
Dans son document, Dr. Calson fait un parallèle entre la «Moore's Law» et les améliorations de productivité dans le séquençage et la synthèse de l'ADN.
|
Cet article analysé par le magazine «The Economist» en 2006, et la «Carlson Curve» fait son apparition dans la littérature. Ainsi est née une courbe pour le monde de la recherche génomique. |
|
|
http://www.kurzweilai.net/images/Carlson(PaceAndProliferation)figure1.gif |
|
Cette courbe est mise à jour chaque année par le «National Human Genome Research Institute». En fait, ils font la mise à jour de deux courbes: La courbe de coût du séquençage pour 1000 paires de bases d'ADN et la courbe des coûts de séquençage pour un génome complet. Ils font la comparaison de ces deux courbes avec la «Moore's Law».
|
|
|
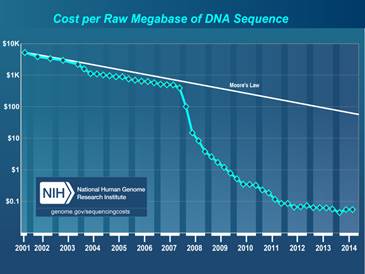
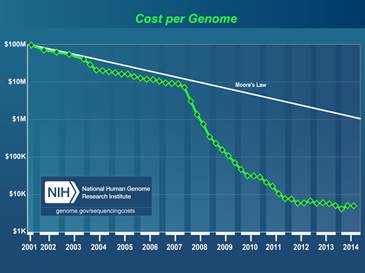
Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP) Available at: www.genome.gov/sequencingcosts. Accessed [21 Fév 2015].
Nous pouvons voir une petite baisse des coûts en 2003-2004, la courbe se détache de la ligne de «Moore's Law» et nous pouvons voir une baisse drastique des coûts la fin 2007. Que c'est-il passé en 2003 et 2007 ? La prochaine figure du «Journal of Pathology Informatics» nous donne une explication.
|
|
|
www.jpathinformatics.org/viewimage.asp?img=JPatholInform_2012_3_1_40_103013_u4.jpg Gullapalli
RR, Desai KV, Santana-Santos L, Kant JA, Becich MJ. Next
generation sequencing in clinical medicine: Challenges and lessons for
pathology and biomedical informatics. J Pathol Inform [serial online] 2012
[cited 2015 Feb 22];3:40. Available from: http://www.jpathinformatics.org/text.asp?2012/3/1/40/103013 |
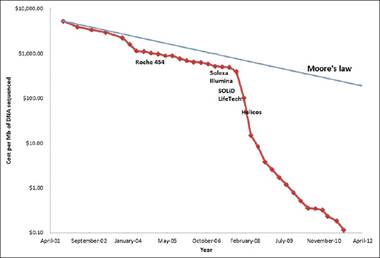
Les pionniers en matière de séquençage, Walter Gilbert et Frederick Sanger, inventent dans les années 70, les premières techniques de séquençage d'ADN rapide. C'est le début de l’ère du séquençage haute vitesse.
Dans les années 90, plusieurs autres méthodes de séquençage de l'ADN se développent dans les laboratoires. Ces méthodes seront utilisées comme techniques pour les générations de séquençage «Next-Generation» qui arrivent à grands pas. ( http://en.wikipedia.org/wiki/DNA_sequencing )
En 2000, la compagnie Lynx Therapeutics publie et commercialise une nouvelle technique de séquençage le "Massively parallel signature sequencing", ou MPSS. Cette technique est la première méthode commerciale disponible de séquençage «Next-Generation». Nous sommes dans la 1ère génération de «Next-Generation Sequencing». ( http://en.wikipedia.org/wiki/DNA_sequencing )
En 2005, la compagnie 454 Life Sciences commercialise le «454 GS20» et nous passons déjà à la 2ième génération « Next-Generation Sequencing » avec cette machine.
D'autres appareils de 2ième arrivent pour concurrencer le 454 de Life Sciences. Vers la mi-2006, Illumina commercialise son appareil, le « Solexa 1G Genetic Analyzer ». En 2007 Applied Biosystems sort son séquenceur, SOLiD (Supported Oligonucleotide Ligation and Detection) et au début 2008, Helicos BioSciences débute la livraison de son appareil HeliScope.
|
|
|
Next-generation
DNA sequencing Jay Shendure & Hanlee Ji, Nature Biotechnology 26, 1135 -
1145 (2008) Published online: 9 October 2008 | doi:10.1038/nbt1486 - http://www.nature.com/nbt/journal/v26/n10/full/nbt1486.html |
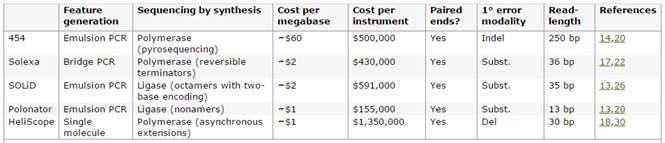
Depuis, nous sommes dans la 3ième génération du « Next-Generation Sequencing ». Les compagnies qui offrent des systèmes sont: Pacific Biosciences avec la technologie SMRT, Oxford Nanopore avec ses biocapteurs à nanopores et Ion Torrent avec sa technologie « Ion PI™ Chip ».
Les produits de deuxième et troisième générations diffèrent dû au fait que la première étape d'amplification d'ADN n'est pas nécessaire. Les brins d'ADN de l'échantillon sont séquencés directement au niveau d'une seule molécule en utilisant des polymérases de protéines modifiées. L'avantage de ces méthodes est d'éviter l'étape d'amplification PCR.
Le site WWW du « National Human Genome Research Institute », dans ses notes explicatives des courbes de coût du séquençage pour 1000 paires de base d'ADN et coût de séquençage pour un génome complet, confirment le changement de technologie séquençage en 2007. Ils passent donc des instruments de première génération à capillaires aux instruments plus rapides de deuxième génération.
|
|
|
Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP) Available at: www.genome.gov/sequencingcosts. Accessed [22 Fév. 2015]. |

Voyons maintenant les différences entre les méthodes d’Electrophorèse à Capillaire Sanger et les méthodes NGS, donc le passage de la première à la deuxième génération de séquençage
2-Principes du NGS Illumina
- Général
- Extraire l'ADN
- Préparation d'ADN
- Générer les regroupements
- Séquencer en Next Generation
2.1 Général
Étapes du Séquençage NextGen
Le principe du séquençage de génome consiste à partir de petits bouts de séquences multiples provenant de plusieurs individus et de reconstruire une méga librairie de 20 000 à 25 000 gènes humains. Comme il existe beaucoup de subtilités et de variantes d'un individu à l'autre, il était essentiel d'inclure un grand nombre d'individus ayant des caractères biologiques variés pour réussir à créer une librairie couvrant tout les gènes humains. Il aura fallu près de 13 ans avec les moyens de ce temps pour y arriver et terminer le 13 avril 2003. (Réf. Jaenish R, 2003)
2.2 Extraire l'ADN
Préparation d'échantillons d'ADN:
*Recommandations importantes pour s'assurer de la réussite du séquençage:
- Mesurer la concentration d'ADN en utilisant le réactif PicoGreen
(Nanodrop ou Spectrophotométrie ont tendance à surestimer la concentration des échantillons ce qui rend la quantité de matériel de départ inappropriée)
- Utiliser de l'eau pure pour resuspendre ou dans du Tris-Hcl 10mM pH 7.5-8.5 (Pas d'EDTA car cela peut bloquer certaines réactions enzymatiques)
- > ou = à 1ug d'ADN / échantillon est requis, dans un volume < ou = à 40ul.
(Réf. Odile Neyret, 2015)
2.3 Préparation d'ADN
- DNA est fragmenté par sonication: Librairie de fragments
- Transformer les extrémités adhésives en coupures franches à l’aide d’exonucléases, y ajouter les groupements phosphates et enfin, ajouter des nucléotides A

- Ligation à deux différents adapteurs: le brin sens avec un type d'adaptateur A, le brin anti-sens avec un adaptateur B.

(Réf. Illumina Sequencing Technology, 2014)
2.4 Générer les Regroupements avec cBOT Cluster Generation
|
Le système: |
|
|
Le système est muni de plaques de verres constitués de microsillons |
|
|
Les sillons sont tapissés avec deux types d’amorces A’ et B’, complémentaires aux adaptateurs A ou B sur les fragments d'ADN |
|
|
Les fragments d'ADN passent dans les sillons des plaques |
|
|
|
|
Extension avec l'ADN polymerase: génération d'ADN complémentaire |
|
|
Le double-hybride formé est dénaturé par chaleur, détachant la molécule originale qui est lavée et éliminée |
|
|
L’extrémité libre comporte l’autre type d'adaptateur (B) qui s’hybride avec l’amorce B’ qui est aussi rattachée à la plaque par lien covalent |
|
|
La polymerase faire un autre brin complémentaire à celui rattaché en pont par ses deux extrémités. On redénature pour séparer les brins néosythétisés. |
|
|
Les cycles d’amplifications des ponts sont répétés jusqu’à ce qu’il y ait une multitude de ponts formés. |
|
|
Les double-brins sont dénaturés. Les brins anti-sens liés à un type d’amorces B sont coupés et on rince pour les éliminer, ne laissant que des brins sens. |
|
|
On ajoute des amorces libres en solution qui vont s’hybrider avec les fragments de DNA pour la partie séquençage proprement dite. |
|




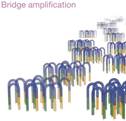

(Réf. Illumina
Sequencing Technology, 2014)
2.5 Séquencer en Next Generation
(NextGen)
Aussi connu sous le nom de ''Séquençage massif en parallèle'' ou ''Massively parallel sequencing''
Comparer NextGen avec Séquençage par Sythèse Sanger:
Séquençage par Synthèse (Sanger)
A) 96 à 384 amplifications de l'échantillon d'ADN sont exécutées.
B) Les amplicons sont séparés par taille par électrophorèse sur gel ou maintenant plus souvent, dans des capillaires.
C) Le séquençage est fait par ordinateur qui fait la détection et l'identification des nucléotides par fluorescence. Chaque type de nucléotide est couplé à une molécule de différente longueur d'onde.
|
|
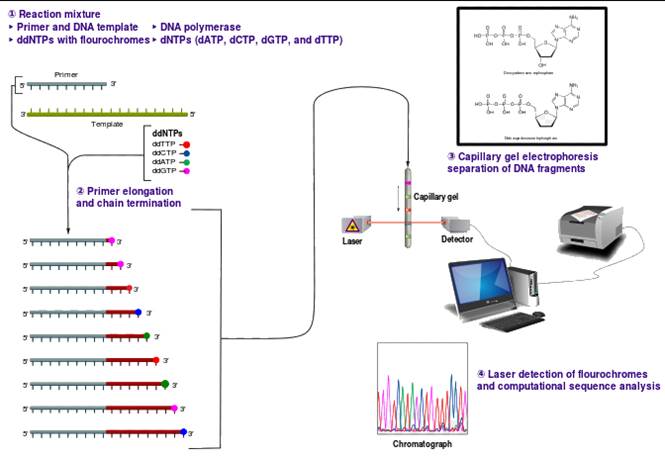
Séquençage par NextGen
Tout d'abord, dès la préparation des échantillons, on épargne du temps par rapport à la méthode par synthèse (Sanger). Le séquençage par NextGen ne nécessite pas de clonage impliquant de sélection clonale qui nécessite quelques jours de préparation et de purification pour obtenir suffisamment de matériel. Ces étapes sont nécessaires car pour le séquençage par synthèse Sanger, il faut posséder une librairie de plusieurs copies pour être capable de détecter le signal.
L'une des grandes différences entre le séquençage par synthèse ou Sanger et NextGen est que le séquençage et la détection du signal se font simultanément pour NextGen. À chaque cycle d'amplification, la détection est faite immédiatement. Il n'y a pas d'étape de migration, ce qui contribue à réduire beaucoup le temps nécessaire à l'exécution du séquençage. Ceci implique aussi que la quantité de matériel séquençable à la fois, n'est plus tributaire de l'espace disponible sur capillaires. Pour le séquençage NextGen, il s'exécute des centaines de millions de réactions en même temps.
Méthode NextGen:
1) Ajout de la polymérase et de nucléotides couplés à des molécules fluorescentes de différentes couleurs pour chaque type de nucléotide
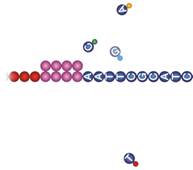
(Réf. Illumina Sequencing Technology, 2014)
2) À chaque cycle d'amplification, le laser excite les fluorophores et détecte quel nucléotide a été rajouté. La détection du signal est rendue possible grâce au fait que chaque signal est envoyé par un Regroupement ou Cluster de fragments qui ont été fait à l'étape Regroupement ou Clustering décrite ci-haut. Plusieurs
Regroupements ou Clusters sont analysés simultanément.
3. Méthodes d'assemblage possibles en Séquençage NextGen
- Séquençage avec Référence
- Séquençage De Novo
3.1 Séquençage avec Référence
Les génomes de plusieurs espèces sont maintenant complétés. Après que le séquençage NextGen soit fait, l'assemblage et alignement des reads pour produire la séquence du résultat final, prend avantage des séquences déjà connues à titre comparatif. Ceci accélère la production et assure une plus grande confiance en les résultats obtenus.
3.2 Séquençage De Novo
Il reste toutefois encore plusieurs génomes sans référence et certaines séquences à obtenir ne sont pas nécessairement connues à l'avance. On doit donc bâtir une séquence De Novo. Comme c'est le cas pour les petits génomes par exemple.
En utilisant des approches via une lecture par les deux extrémités de chaque fragment, on optimise la couverture de lecture de chacun des fragments qui n'aurait été parfois que partielle en faisant un séquençage par un seul bout.
L'idéal est de combiner deux approches. On utilise des fragmentations longues qui servent de ''référence'' pour aligner les petits fragments parfois difficiles à aligner sans canevas de base. Comme les petits fragments peuvent être lus avec une meilleure Résolution, ils permettent de venir remplir les espaces incomplets laissés dans les grands fragments. Les longs fragments étant lus avec moins de Résolution, il arrive souvent qu'ils comportent des sections de moindre qualité. Donc la combinaison des deux types de lectures donne d'excellents résultats. C'est une bonne approche pour des séquences ayant des éléments répétitifs par exemple.
(Réf.: Li R et al, 2010; Ruiqiang Li et al., 2012)
4-NGS: Adaptabilité
aux besoins
▪ Résolution ou Coverage
▪ Exemples d'applications, Génome entier, Petits génomes
4.1 Résolution ou Coverage
La Résolution réfère au nombre moyen des reads produits pour aligner et reconstruire la séquence. Par exemple une Résolution de 30X veut dire que chaque base aura été retrouvée en moyenne dans 30 reads.
4.2 Exemples d'Applications, Génome Entier, Petits Génomes
4.2.1 Application pour la Résolution de haut niveau comme
1000X
En cancérologie, les mutations des cellules cancéreuses se retrouvent dans de faibles proportions à travers un grand groupe de cellules. Avec des hauts niveaux de Résolution de 1000X, NextGen permet la détection de ce type de mutations, ce qui ne serait pas possible avec la méthode Sanger.
La Résolution haut niveau permet aussi de quantifier des différences subtiles de changements d'expression avec d'ARN qui ne pourraient pas être possibles avec le microarray.
4.2.2 Application pour la Résolution de niveau 30X
Avec une Résolution de 30X, on peut séquencer le génome entier en quelques jours. Ce qui a pris des années avec la méthode Sanger. Ceci implique aussi évidemment un coût beaucoup moindre avec NextGen.
4.2.3 Application pour la Résolution de niveau comme 10X
NextGen à basse résolution a pour avantage majeur, une grande rapidité et un faible coût. Ce qui peut être spécialement intéressant pour pouvoir réagir rapidement en face d'infections bactériennes. Par exemple, il a été possible en Europe de séquencer rapidement une souche dérivant d'E.coli permettant une meilleure compréhension rapide de la virulence de cette bactérie. Ceci a permis aux autorités de réagir rapidement, évitant une potentielle pandémie.
La Résolution de faible niveau est aussi très utilisée pour découvrir des variantes génomiques car son faible coût permet facilement de pouvoir couvrir des larges spectres de séquençage.
(Réf.: Mardis E., 2011 et Illumina, 2011)
5. Comparaison entre
les trois générations de séquençage.
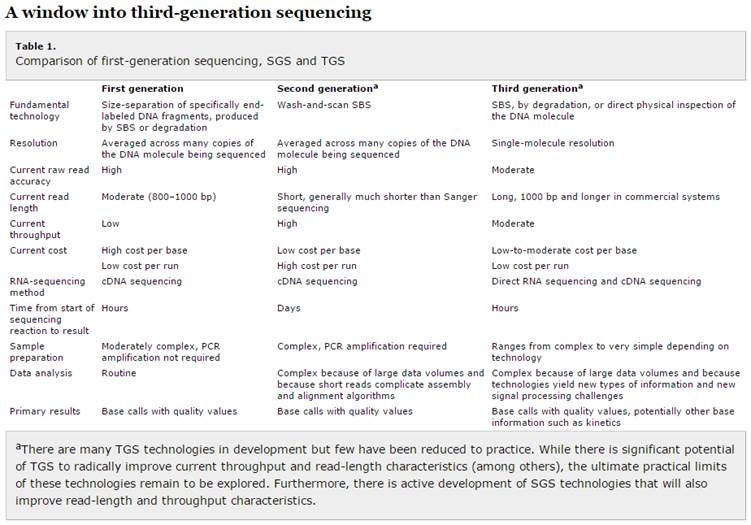
A
window into third-generation sequencing, Eric E. Schadt*, Steve Turner and
Andrew Kasarskis, Human Molecular Genetics Volume 19, Issue R2Pp. R227-R240,
September 17th, 2010. http://hmg.oxfordjournals.org/content/19/R2/R227.full
Nous avons contacté les centres suivants pour avoir des données comparatives des divers équipements de ces centres de recherches en génomique.
- le Centre d'innovation Génome Québec
- l'IRIC
- l’Institut de recherches cliniques de Montréal
|
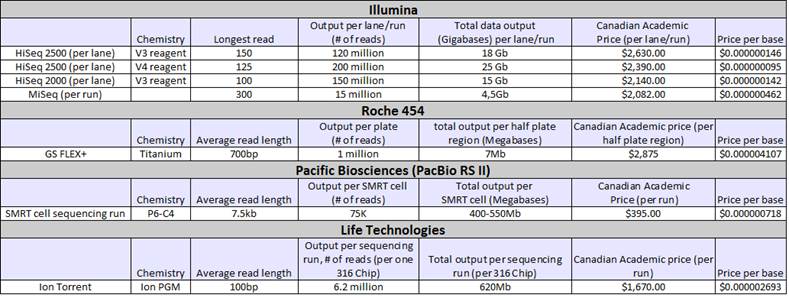
Sharen Roland, Gestionnaire de clients, du Centre d'innovation Génome Québec nous a donné les données pour remplir le tableau suivant. Nous pouvons voir que ce centre possède des équipements de deuxième et troisième génération de séquençage. |
|
|
|
Raphaëlle Lambert, coordonnatrice de la plateforme génomique de l'IRIC, nous a fait parvenir les données suivantes: |
|
|

|
Odile Neyret, responsable d'équipe, du plateau Biologie moléculaire et génomique fonctionnelle de l'IRCM nous donne ceci pour le tableau : |
|
|

{kind=link}
figure1.gif){kind=link}
{kind=link}
6. Séquençage NextGen d'ADN Bisulfité:
Étudier la Méthylation d'ADN au niveau du nucléotide individuel, sur génome entier
- Méthylation du génome
- Représentation Réduite et Séquençage Bisulfité (RRSB)
6.1 Méthylation du génome
6.1.1 Méthylation du génome: Général
La méthylation est une modification épigénétique, c'est-à-dire que c'est de l'information qui se transmet de façon héréditaire mais qui n'est pas encodée par la séquence en nucléotides. Chez les mammifères, la méthylation a plusieurs fonctions comme la régulation de l'expression des gènes, l'imprinting et l'inactivation du chromosome X. La méthylation est aussi importante car elle augmente la stabilité génomique et protège contre les mécanismes de transpositions d'éléments.
(Réf.: Meissner J. et al. 2005 et Gu H, et al, 2011)
6.1.2 Méthylation du génome: Pourquoi l'étudier?
La méthylation est un phénomène d'extinction génique. La méthylation empêche la liaison des complexes enzymatiques et prévient la transcription.Les groupements méthyles sont souvent sur des séquences dites CpG1 ou îlots CG souvent associés aux promoteurs et donc à l'activité du gène en position 1. La méthylation est aussi liée aux facteurs environnementaux. Par exemple la nutrition peut affecter le taux de méthylation d'ADN. Il a aussi été montré que la présence de cocaïne ou d'alcool peut influencer le taux de méthylation.
(Réf.: Meissner J. et al. 2005 et Gu H, et al, 2011; Vaillancourt et al., 2014)
6.2 Représentation Réduite et Séquençage Bisulfité (RRSB)
6.2.1 Principes de la RRSB
La méthode RRSB a pour but de réduire la quantité de fragments à séquencer pour ne garder que les séquences d'intérêt ayant des méthylations. Ainsi les coûts et le temps de séquençage et d'analyse sont beaucoup réduits. RRSB est une approche qui a une résolution au niveau du nucléotide individuel qui est très sensible et qui produit une mesure quantitative d'ADN méthylé. Elle est adéquate pour analyser la méthylation sur un génome entier. La méthode de traitement d'ADN au bisulfite permet de transformer chimiquement les C non méthylés en Uracil et les C-méthylés sont protégés du changement, laissant alors des C seulement aux endroits où les C étaient méthylés.
(Réf.: Chatterjee A, et al. 2012.; Bell
Jtet al., 2011)
6.2.2 Méthode RRSB

(Réf. Babraham
Bioinformatics , 2013)
1- Digestion enzymatique :
Digestion enzymatique indépendante de l'état de méthylation des CpG. L'enzyme de restriction Msp1 reconnaît tout motif CCGG sur DNS double brin et coupe le pont phosphodiester, laissant des extrémités à bouts cohésifs = C/ CGG ; GGC / C
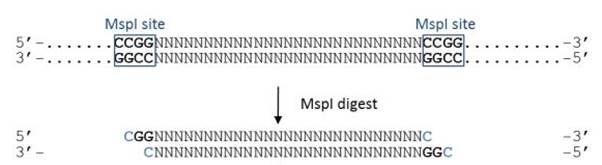
(Réf. Babraham
Bioinformatics , 2013)
2-Remplissage et addition de A aux extrémités:
Les extrémités sont remplies à l'aide de l'enzyme fragment de Klenow et un excès de dATPs sont ajoutés en solution pour s'assurer d'obtenir des extrémités à bouts francs et ayant un A à la fin.
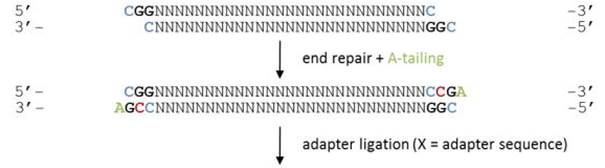
(Réf. Babraham
Bioinformatics , 2013)
3- Adapteurs méthylés:
Des adaptateurs méthylés sont ajoutés aux fragments d'ADN pour servir d'amorces à l'étape d'amplification. Les C sont méthylés pour qu'ils ne soient pas convertis lors de l'étape de méthylation.
4-Purification des fragments:
Les fragments de DNA de 40 à 220 paires de bases représentent la majorité des îlots de CpG.
5-Conversion Bisulfite:
Les C non méthylés sont transformés chimiquement en U. Les C méthylés restent des C méthylés.
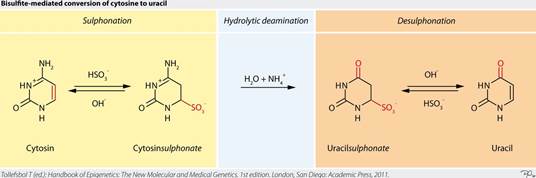
6-Amplification PCR:
Les fragments d'ADN sont amplifiés avec des amorces complémentaires aux adaptateurs.
7-Purification:
Les réactifs en solution sont éliminés pour ne garder que les fragments d'ADN d'intérêt.
8-Séquençage NextGen
9-Alignement et Analyse des Séquences
obtenues
6.1.3 Étude Éipgénétique:
Projet Dépendance à la cocaïne
RRBS aura permis de réaliser un projet d'étude mené par l'équipe Vaillancourt K. et al en 2014. Ils ont démontré une différence de méthylation entre des cerveaux de patients ayant fait usage régulièrement de cocaïne versus des cerveaux de patients n'ayant pas consommé de cocaïne, dans les régions du Caudate et du Nuleus Accumbens. Le séquençage NextGen sur RNA-seq a été fait sur des échantillons issus des mêmes cerveaux, ce qui a permis de confirmer les résultats obtenus.
7. Réflexions et Conclusion
Les chromosomes font un excellent travail en contenant tout notre génome dans un si petit espace. Après avoir découvert tant de choses, les chercheurs de ce monde sont pris au piège pour emmagasiner toute cette information. Car contrairement aux chromosomes, nous n'avons pas autant d'années d'expérience en gestion d'inventaires d'information.
Le NGS nous aide à percer bien des secrets, mais soulève aussi d'autres questions. Plus la technologie NGS avance, plus les coûts baissent et la quantité des données augmente. Par exemple, l'Illumina 2500 génère des fichiers de 25Gb. Beaucoup de collaborations se font entre chercheurs de différents continents des nos jours. Mais comment transférer ces quantités de données rapidement, sans erreur et d'une manière sécuritaire? Comment protéger ces donnés de recherches contre l’espionnage industrielle? Dans ce monde de profits et de retour d'investissement, des années de recherches peuvent être volées avec une simple clef USB.
Il y a beaucoup d'autres questions à se
poser avec ces données qui arrivent en tsunami mais, concentrons nous sur ce
que ces données peuvent nous apporter pour l'avancement des recherches et
finalement trouver des solutions aux maladies qui nous affectent. D'autres
sciences, telles la sécurité informatique seront nécessaires pour nous aider à
conserver l'ordre et le respect de la propriété intellectuelle.
8. Références:
-Odile Neyret, 2015. Responsable d'équipe, du plateau Biologie moléculaire et génomique fonctionnelle de l'IRCM.
-Illumina 2014,
Sequencing Technology.
-Li R et al, 2010.
Nature: 463:311-317. The sequence and de Novo assembly of the giant panda
genome.
-Illumina 2011. An
Introduction to Next Generation Sequencing Technology.
-Gu H, et al, 2011. Nat Protoc. 6(4):468-81. doi: 10.1038/nprot.2010.190.
"Preparation of reduced representation bisulfite sequencing libraries for
genome-scale DNA methylation profiling."
-Meissner J. et al.
2005. Nucleic Acids Res. 33(18): 5868–5877. Reduced representation bisulfite
sequencing for comparative high-resolution DNA methylation analysis.
-Babraham Bioinformatics
, 2013. Reduced Representation Bisulfite Sequencing for Methylation Analysis
Preparing Samples for the Illumina Sequencing Platform.
-Mardis E., 2011, Nature, 470:198-203.
-Vaillancourt K. et al (Alpha Diallo) 2014.
Society for Neuroscience. DNA
Methylation in the Striatum of Individuals with Chronic Cocaine.
-Chatterjee A, et al,
2012. Comparison of alignment software for genome-wide bisulphite sequence
data. Nucleic Acids Research40(10): e79
-Ruiqiang Li et al.,
2012, Genome Research 0:265–272. De novo assembly of human genomes with
massively parallel short read sequencing
-Bell Jtet al., 2011.
Genome Biology 12 (1): R10. DNA methylation patterns associate with genetic and
gene expression variation in HapMap cell lines.