In silico reconstruction of an ancestral mammalian genome

Conférencier: Mathieu Blanchette

INTRODUCTION
Les recherches en bioinformatique accordent beaucoup d’importance sur l’annotation génomique fonctionnelle. Depuis quelques années, de plus en plus d’espèces sont séquencés en totalité ou en grande partie de leur région génomique. Maintenant on cherche à savoir d’avantage sur les rôles de chaque région du génome mais surtout comment était leur mode d’évolution.
Nous savons tous que les espèces évoluent soit par un événement de mutations : substitution, insertion, suppression, soit par réarrangement génomique ou autres type de sélections/mutations. Ce travail dont nous allons soumettre un rapport traite uniquement les mutations le plus fréquent: INDEL et ce pour le cas des mammifères. L’idée est de reconstruire les séquences de nucléotide des ancêtres commun de 20 espèces en se basant sur ce qu’on a les concernant.
I-Historique des études connexes :
Du maximum de vraisemblance des algorithmes pour la reconstruction des acides aminés ancestral ou des bases de l'ADN ont été développés et utilisé par plusieurs groupes (Yang et al. 1995; Koshi et Goldstein 1996, Cunningham et al. 1998; Schultz et al. 1996; Pupko et al. 2000, 2002).
En 1997, Zhang et Nei pensent que l’approche maximale de vraisemblance fonctionne mieux que les méthodes de parcimonie pour ce genre de projet. Les méthodes Bayésiens qui tiennent compte des incertitudes dans l'arbre, les longueurs des branches, et les paramètres du modèle ont également été explorées (Schultz et Churchill, 1999; Huelsenbeck et Bollback 2001), bien que il s'agit d’un traitement plus couteuse en méthodes d'échantillonnage Markov Chain Monte Carlo.
À quelques exceptions près (Hein 1989; Fredslund et al. 2003), les algorithmes ont été limitées à une pure substitution de modèles, sans tenir compte des insertions et des suppressions.

b) Méthodes de comparaison:
1. Comparaison statique de séquences simulées avec l’actuelle séquence
Pour assurer une similarité avec celui du locus CFTR, une comparaison des résultats des alignements multiples des séquences de CFTR et des séquences simulés s’imposent. Ceci se fait par l’étude des propriétés du « pair-wise » alignement entre 2 espèces.
Le nombre de substitutions par site selon le modèle Kimura 2-paramètre est résumé sur le Tableau 2. Nous pouvons voir que les séquences simulés et les séquences actuelles sont assez proches dans la partie supérieur du diagonale, sauf pour la souris où les valeurs étaient sous estimés. Dans la partie en bas de la diagonale se trouve les fractions des bases non-répétitives qui ont été alignées
Les simulations comprenaient une insertion des transposons spécifique de la lignée et une augmentation du taux de substitution en CpG des dinucleotides. Pour chaque paire de séquences orthologues généré, nous avons vérifié que le nombre moyen de substitutions, insertions, et des suppressions sont proches de ceux observés dans l'évolution neutre
régions de la grande région de CFTR. Nous avons également vérifié que les distributions des tailles des insertions et suppressions, ainsi que la fréquence et la répartition par âge de chaque type d'élément répétitif sont proches de celles précédemment (IHGSC 2001; IMGSC 2002). De plus amples détails sur le processus de simulation et de sa validation
sont donnés dans les méthodes et Blanchette et al. (2004).
2. Repérage des contenus répétitive
Dans cette partie, on utilise RepeatMasker pour repérer les séquences répétitives évaluées la distribution pour les séquences humaines. L’objectif est d’avoir une moyenne en %divergence, %deletions and %insertions avec certaines familles répétitive.
L’alignement multiple de RepeatMasker est ensuite envoyé à un algorithme glouton qui vise à expliquer le reste des indels. L'algorithme considère que l'alignement est suit l’arbre phylogénétique, c'est-à-dire, que les deux bases sont alignés si et seulement si elles proviennent d'une base commune ancestrale.
L’algorithme sélectionne d’une manière itérative les insertions et les suppressions le long de l’arbre sur plusieurs colonnes d’alignements.
Les coûts sont définis heuristiquement. Le coût d'une suppression est donné par 1 + 0,01 log (L) _0.01 b, où L est la longueur de la suppression et b est la longueur de la branche sur laquelle se cible. Le coût d'une insertion est donné par 1 + 0,01 log (L) _0.01 b_r, L et b sont définis comme ci-dessus et r est un terme qui prend la valeur 0.5 si la répétition du contenu du segment inséré est> 90%.
Si on arrive à avoir une meilleure insertion ou suppression, les gaps seront marqués comme "expliqué".
Enfin, les heuristiques sont utilisées afin de réduire les erreurs dues à un mauvais alignement, notamment en vue de réduire les problèmes causés par deux régions répétitives de deux espèces lointaine alignés par erreur les uns aux autres, et avec d'autres espèces ayant aussi des gaps dans cette région.
Après avoir trouvé quelles positions de l'alignement multiple correspondent à des bases de l'ancêtre, il faut repérer les nucléotides qui était présent à chaque position dans l'ancêtre à l'aide d’une approche de probabilité postérieure standard (Yang et al. 1995)
Les longueur des branches sont inférées avec PHYML (Guindon et Gascuel 2003).
II-3 Analyses et interprétations
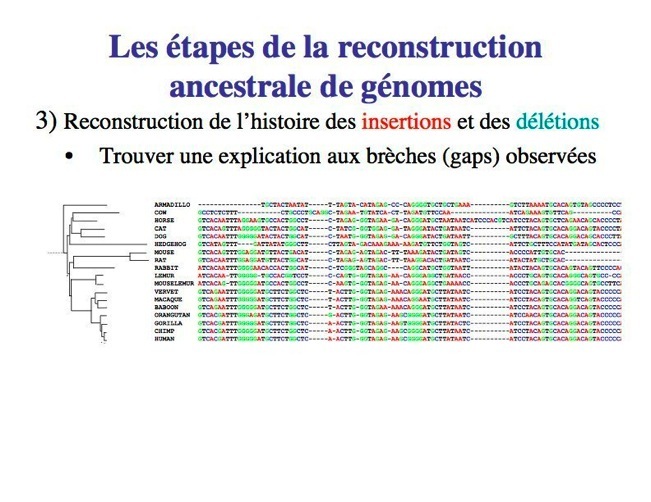
Vu que l'alignement traite des séquences orthologues d'ADN et qu’on a un arbre phylogénétique de ces séquences, le problème de la reconstruction d'un scénario plus parcimonieux des insertions et des suppressions s'explique avec les gaps observés dans l'alignement. Ce problème, appelé Indel du problème parcimonie constitue un élément majeur dans la reconstruction du génome ancestral, et sa solution fournit une information précieuse pour de nombreuses approches d’annotation génomique fonctionnelle.
C’est un problème NP-complet. L’algorithme proposé par Blanchette et. Al est basé sur une programmation linéaire. Il est rapide et donne des solutions assez correctes. L’approche diviser-pour-régner a été choisi afin de pouvoir résoudre une très grande quantité de données, sur une simple machine de bureau, tout en conservant la garantie d'optimalité. Les algorithmes ont été testés sur un ensemble de 1,8 Mb de séquences orthologues des mammifères dans la région CFTR et ont montré des résultats efficaces et précises [1].
- Discussion
Un des résultats non intuitifs de cette étude est l'observation que beaucoup sont les anciens ancêtres dont les génomes peuvent être reconstruits avec plus de précision que ceux de leurs plus récents descendants.
Ceci s’explique dans le cas où l'arbre présente une topologie en étoile, dans lequel chacune des espèces modernes découle directement de l'ancêtre d'une branche autonome, alors il est clair que la méthode de maximum de vraisemblance et la méthode bayésienne
a posteriori des reconstitutions de caractère consensus ancestral fournissent le plus souvent ceux observés dans les n modernes espèces. Plus on a des espèces orthologues, plus on aura la chance de prédire une séquences consensus plus précis. La probabilité d'une erreur dans la reconstruction est:
qui est au plus [4p (1 _ p)] n / 2 (Hoeffding 1963; Le Cam [5 Lemma p.479] 1986). Cette valeur d’erreur approche le zéro rapidement d’une manière exponentielle quand n augmente. Lorsque n est trop petit, l'ancêtre n'est probablement pas reconstructible (Mossel 2003).
-Les limitations importantes des simulations
(1)Les taux de substitutions, suppressions, et les petites insertions sont supposés être constants dans la position des séquences et à travers les branches homogènes (Eizirik et al. 2001).
(2)Divers modes de sélection ne sont pas modélisés, y compris les types de purification spécifiques de sélection dans les codons et dans les autres régions, et la sélection positive pour les nouvelles fonctions.
Ces sélections (selon le cas) pourront aider ou pas à la reconstruction ancestrale.
(3)Certaines mutations de nucléotides au niveau des processus tels que l'ADN polymérase (Nishizawa et Nishizawa 2002) ou de conversion génique ne sont pas incluses dans la simulation. Alors que ces dernières pourraient changer les schémas de l'évolution moléculaire, dans certaines régions.
(4)les processus mutationnel à tandem et segmentaire : duplication, inversion, et le réarrangement ne sont pas incluses dans les simulations. En effet, la région du protéine CFTR, indique une pénurie de tels changements. Toutefois, les réarrangements ne sont pas des facteurs à négliger car ils jouent aussi dans l’évolution des espèces en termes de séquence. On estime que 10% de la euchromatique génome humain a fait l'objet de récentes duplications (Samonte et Eichler 2002) et / ou un excès de réarrangements (Kent et al. 2003; Pevzner et Tesler 2003).
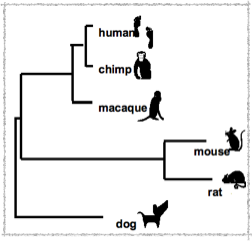
Avec les séquences génomiques existantes à partir du 19 mammifères, 1.1 Mb de l'ancienne séquence du génome du locus CFTR a été reconstruit. Ainsi, il est possible de prédire l' histoire évolutif des bases de la région reconstruit, en se basant sur la quantité de l'ADN, qui varie selon qu’il y a de l'insertion, du suppression, et de substitution dans les différentes lignées de mammifères placentaires depuis l'ancêtre commun, montrant des variations considérables entre les lignages.
Dans les années à venir, telles reconstructions pourront aider à identifier et à comprendre les caractéristiques génétiques communes à eutherian de mammifères et faire la lumière sur l'évolution de l'homme ou des primates.
Dans les années à venir, telles reconstructions pourront aider à identifier et à comprendre les caractéristiques génétiques communes à eutherian de mammifères et faire la lumière sur l'évolution de l'homme ou des primates.
Les longues périodes de conservation d’une région suivi d’un changement brusque fourniront des indices sur l'évolution des nouvelles traits de l'homme (Goodman et al. 1971; Challem 1997; Enard et al. 2002). Tous les efforts dépendent des chercheurs, directement ou indirectement, sur la reconstruction de l'histoire évolutive des bases du génome de l’homme à partir de ceux des génomes de nos ancêtres lointains.
La séquence reconstruite ancestral peut également être utilisé pour recueillir des statistiques sur les taux de gain et de perte de l'ADN dans les différents lignages eutherian [2].
AMÉLIORATIONS
III- Les continuités du travail
Le laboratoire de David Haussler a travaillé en mai 2004 sur l'évolution moléculaire du génome humain, l'intégration inter-espèces et comparative de génomique pour étudier la structure des gènes, les fonctions. Pour se faire, ils ont élaboré un algorithme combiné avec des méthodes statistiques toute en se concentrant sur l'analyse et la classification de calcul de l'ADN, l'ARN, et de séquences de protéines. Haussler avait pu remarquer l'existence des régions ultra-conservés depuis des millions d'années dans le génome humain en Mai 6, 2004 et conclu que ces régions codent probablement pour des fonctions importantes. Son travail est complémentaire avec celle de Blanchette et al. vu qu’il tiens en compte les protéines dans ses études [3].
Le groupe a également identifié des segments du génome qui ont subi des changements très rapides dans une même espèce, comme un gène lié au développement du cerveau qui a changé de façon spectaculaire entre chimpanzés et humains Nature, Aug 16, 2006.
Des études plus élargis ont été fait en 2008 sur les zones conservés à 100% d'identité, sans insertions ou suppressions, entre les régions orthologues des génomes de l'homme, du rat et de la souris, mais cette fois-ci sur les poulets et autres. Presque tous ces segments sont également conservés dans les génomes du poulet et du chien, avec une moyenne de 95 et 99%. Plus souvent situées dans à proximité de gènes impliqués dans la régulation de la transcription et de développement [3]. En générale ces sont des protéines qui semblent être essentiels pour l'ontogénie des mammifères et autres vertébrés.
Grace aux résultats de séquençage de l’ancêtre commun de l’homme et de la souris, l’UC Santa Cruz a pu expliquer en juin 2006 l’existence d’un réarrangement chromosomique au niveau du chromosome 2 de leur ancêtre.
En effet, le génome de la souris cartographié sur le génome humain a montré les 34% de cas identiques, indiquant l’existence des réarrangements des deux génomes depuis leur dernier ancêtre commun, il y a environ 75 millions années.
RÉFÉRENCES
[1] L. Chindelevitch, Z. Li, E. Blais, and M. Blanchette. On the Inference of Parsimonious Indel
Evolutionary Scenarios, Journal of Bioinformatics and Computational Biology, 4(3): 721–744 (2006)
[2] M. Blanchette, E. D. Green, W. Miller, and D. Haussler. Reconstructing large regions of an ancestral
mammalian genome in silico, Genome Research, 14(12): 2412–2423 (2004)
[3] G.Bejerano, M. Pheasant, I. Makunin, S. Stephen, W. J Kent, J. S. Mattick, D. Haussler
Ultraconserved Elements in the Human Genome, Science Express Vol. 304. no. 5675, pp. 1321 – 1325
on 6 May 2004
[4] T. Speed and H. Huang (Eds.): RECOMB 2007, LNBI 4453, pp. 196–210, 2007.c_Springer-Verlag
Berlin Heidelberg 2007
[5] B. Diallo, V. Makarenkov, and M. Blanchette. Exact and Heuristic Algorithms for the Indel Maximum
Likelihood Problem, Journal of Computational Biology, 14(4): 446–461 (2007)
[6] J. Felsenstein and G. A. Churchill. A Hidden Markov Model approach to variation among sites in rate
of evolution, Molecular Biology and Evolution, 13(1): 93–104 (1996)
[7] J. Fredslund, J. Hein, and T. Scharling. A Large Version of the Small Parsimony Pro- blem. In WABI,
volume 2812, pages 417–432 (2003)
[8] L. Chindelevitch, Z. Li, E. Blais, and M. Blanchette. On the Inference of Parsimonious Indel
Evolutionary Scenarios, Journal of Bioinformatics and Computational Biology, 4(3): 721–744 (2006)
[9] G. Tesler. GRIMM: genome rearrangements web server, Bioinformatics 18(3): 492-493 (2002)
[10] S. Schwartz, W. James Kent, A. Smit, Z. Zhang, R. Baertsch, R. C. Hardison, D. Haussler, and W.
Miller Human-Mouse Alignments with BLASTZ, Genome Research, 13: 103-107 (2003)

CONCLUSION
Les mammifères de nos jours sont les résultats d’une série de changement depuis l’ère Crétacé-Tertiaire. L’arbre phylogeny de ces espèces facilite la reconstruction de la séquence génome de leur ancêtre commun. En effet, grâce aux ressources disponibles actuellement, il est possible d’espérer par exemple à avoir jusqu’à 98% des bases en reconstruction sur une échelle de mégabase, les régions euchromatique d'un génome ancestral de l’eutherian à partir de quelques 20 génomes optimale choisi mammifères modernes.
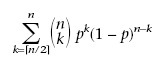
MÉTHODE
II- Le travail de Blanchette et al 2004
II.1- Problématique et intérêts
On a peut être les génomes complets de certaines espèces mais il reste encore beaucoup de mystère à percer qui pourra jouer sur la connaissance de nos futures, de mieux se préparer aux divers risques qui se présentent et de mieux combattre les anomalies.
L’intérêt à ce projet est qu’il permet de regarder comment les génomes ont évolué en cherchant le génome de l’ancêtre commun et par la suite on pourra analyser cela, ce qui conduira aux annotations des différent régions génomique pour aboutir à une meilleure compréhension de notre génome.
II.2- Procédures
a) Procédures d’identification des insertions et des délétions
En vue de reconstruire le génome ancestral, la procédure prend en entrée des séquences génomique de quelques mammifères avec une arbre phylogénétique pour trouver un peu de tout du génome ancestrale à savoir ses fonctions, les régions introns ou intergénique, les séquences répétés, les régions dépourvus de fonctions, etc.…
Operations mutationnelles
•Substitutions, suppressions, insertions
1.Identification des régions synteniques pour chaque espèce
L’identification des régions orthologues se fait par Blastz (Blastz,Schwartz et al 2003), puis les blocs de synténie seront ordonnés par un programme de chaînage et de compensations :Chaining/netting program (Kent et al.) ou sur (GRIMM,Tesler 2002). Pour finir, les cases encodées vont suivre un séquençage ciblé par BAC
2.Alignement multiple des génomes
Une première étape cruciale vers la reconstruction des séquences ancestrales est de construire un alignement multiple exact des séquences existants, établissant ainsi des relations entre les orthologues et à chaque séquence de nucléotides. TBA (Blanchette et al. 2004) est utilisé en tant qu’outil d'alignement. Elle est basé sur le programme pair-wise alignement de BLASTZ (Schwartz et al. 2003).
L’arbre phylogénétique correspondant aux séquences est ensuite déduit en utilisant le modèle HKY (Hasegawa et al. 1985) et aussi avec le programme PHYML (Guindon et Gascuel 2003).
Comme paramètre: Ts/Tv = 2, p(a) = p(t) = 0.3, et p(c) = p(g) = 0.2, sauf que le taux de substitution de paires CpG sont 10 fois plus élevé que les autres taux (Siepel Haussler et 2003). Les suppressions sont initialisés à un taux d'environ 0.056 fois le taux de substitution, leur longueur est choisie en fonction d'une distribution empirique précédemment (Kent et al. 2003).
Blanchette et al. a prédit quelles positions de l’alignement correspondant aux bases ancestrales qui correspondent aux nucléotides insérés après l'ancêtre. Un algorithme glouton se charge de chercher à expliquer le fait observer l'alignement l'aide d'une série d'insertions et suppressions de maximum de vraisemblance (voir méthodes de comparaison).
3.Reconstruction des historiques insertion/suppression
Cette étape est juste une simulation qui consiste à trouver la plus probable explication pour les gaps observées. En d’autre terme, cela définit la présence ou l'absence d'une base à chaque position de chaque ancêtre.
4.Inférer en maximum de vraisemblance les nucléotides à chaque position
Les séquences Ancestral seront inférées.
La précision de la reconstruction dépend essentiellement de la longueur du début des branches. Autres simulations ont révélé que si les grandes lignées placentaire a divergé instantanément (au début de branches de longueur égale à zéro), nous serions en mesure de reconstruire la simulation de la séquence ancestral Boreoeutherian, y compris les régions répétitive, avec moins de 1% d'erreur.
En revanche, si les longueurs des branches déduites par Eizirik et al. (2001) s'est avéré à sous-estimer les longueurs par un facteur de deux, le taux d'erreur passe à 3%, et à 6% si elles ont été sous-estimée par un facteur de quatre.
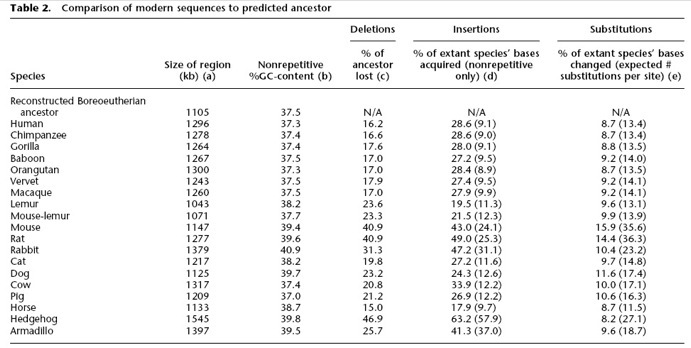
Tableau 1. Comparaison des séquences
Dans ce tableau, une liste de certaines propriétés des séquences des espèces qui existent dans la grande-locus CFTR et les prévisions des changements engagés au cours de l'évolution de la séquence ancestrale Boreoeutherian.
(a) Longueur de la séquence.
(b) Fraction de bases non-répétitive qui sont des G ou des C.
(c) Deletions: pourcentage de la séquence ancestrale perdue dans chaque espèce.
(d) Insertions: pourcentage des séquences des espèces existantes qui a été inséré depuis l'ancêtre reconstruit.
À noter que, le pourcentage des séquences des espèces existantes résulte de l’insertion de séquences non-répétitive, en utilisant RepeatMasker pour identifier les séquences répétitives.) La fraction des bases non-répétitive inséré chez le lapin et le hérisson est très probablement dû au manque de bibliothèques dans RepeatMasker pour les transposons spécifiques à ces espèces.
(e) Substitutions: pourcentage des bases des espèces existantes dérivées d'une base ancestrale spécifique, mais se diffèrent à partir de cette base (ce qui est différent de la mesure de l'identité du pourcentage standard, où seuls les bases alignés sont considérés).
Entre parenthèses, le nombre de substitutions par site dans le cadre d'un modèle Kimura 2-paramètres (Kimura, 1980) est donnée, ici en utilisant seulement les bases alignées.
ÉTAPES
Pris de la présentation: Algorithme pour la reconstruction informatique du scénario d’évolution le plus vraisemblable Réalisée par: Abdoulaye Baniré Diallo.



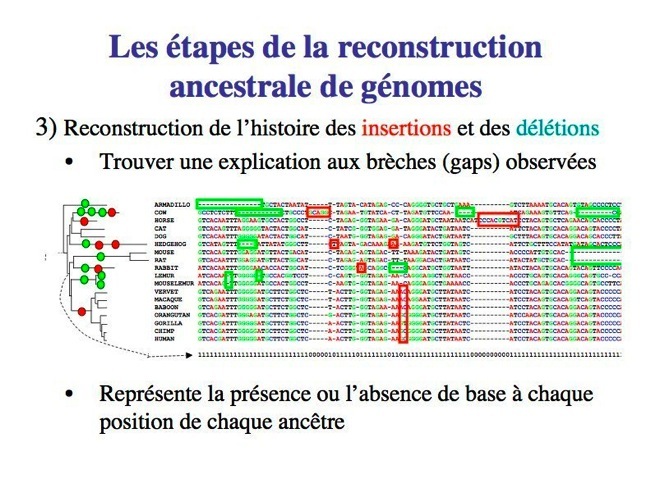
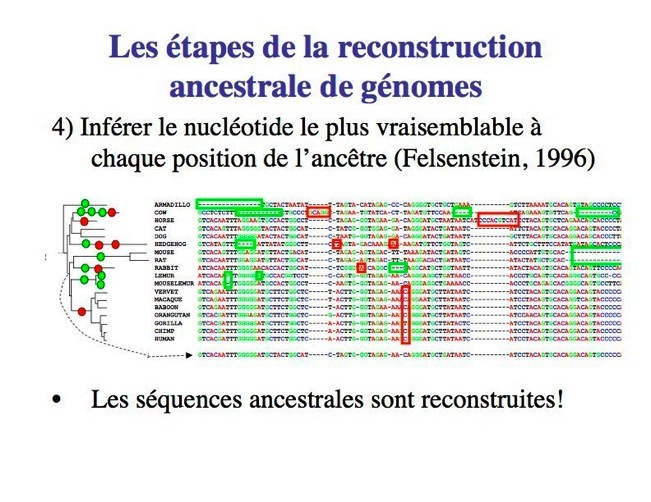
Exemple de reconstruction d'une séquence ancestrale Boreoeutherian basé sur des séquences orthologues dérivées d'une MER20 rétro-transposon.
Les flèches indiquent les positions où les ancêtres reconstitués sont différents de la MER20 consensus. Les flèches longues indiquent les positions où la connaissance de la séquence consensus MER20 aurait changé la prédiction des bases ancestrales. La position de la séquence de l'homme est visible au chr7 :115,739,755-115, 739.899 (NCBI build 34). L'alignement de l'accompagnement non-répétitif d’ADN (données non présentées) vérifie que les séquences de différentes espèces sont en fait orthologues. Les arbres et les branches sont directement tirés d’Eizirik et al. (2001).
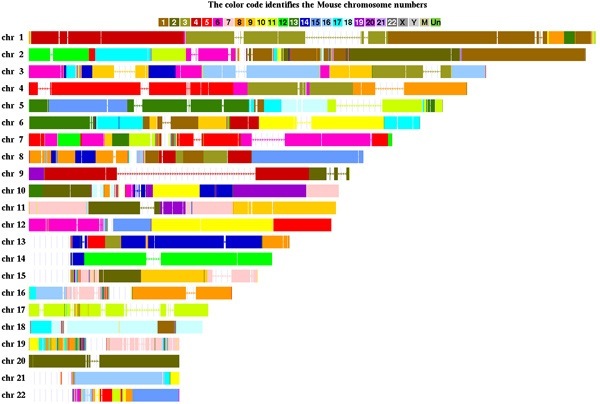
Figure 2. Comparaison entre les génomes de l’homme et de la souris
Source: Genome Browser at UC Santa Cruz by Hiram Clawson and Kate Rosenbloom. 09 June 2006
La cohérence de la couleur indication démontre l'étroite identité (95%) entre les deux génomes depuis leur dernier ancêtre commun, à environ 5 millions d'années. Autrefois nommé chr12 et chr13, renommés en chr2a et chr2 du fait qu'ils partagent une origine commune avec chr2 de l'homme. Ici, ils sont représentés par la même couleur
T. Speed and H. Huang ont sortie en 2007 un article intitulé “Reconstructing the Phylogeny of Mobile Elements”. Cet article présente une méthode qui prend en compte les réarrangements génomiques pour expliquer le mécanisme et l’histoire évolutive des espèces en travaillant sur les éléments mobiles et les régions répétitifs [4]. Ceci est complémentaire au travail de Blanchette et al.
La même année, Abdoullayé Diallo a travaillé un algorithme heuristique et exact utilisant la méthode de maximum de vraisemblance traitant ainsi les indels non chevauchants. À l’heure actuel, un projet est en cours afin d’améliorer le modèle de Diallo et al 2007 pour trouver le scénario d'évolution le plus vraisemblable incluant indels et substitutions [5].
Séminaire de Bioinformatique - UQÀM - Hiver 2009 - Page Web: Pablo Eduardo Moreno et Nary Raveloson
