Séminaire
en bioinformatique (BIF7002)
Rapport
Régulation des GTPases Ras :
De la génomique intégrative à l’identification de cibles thérapeutiques
|
Rédactrice |
Amal
Louhibi |
|
Date
de remise |
19-03-2008 |
|
Groupe |
20 |
|
Professeur |
Vladimir
Makarenkov |
Table des matières
i) Introduction
1.
Biologie des
systèmes et génomique fonctionnelle
2.
Démarche suivie dans
la biologie des systèmes
3.
Les petites
protéines G
4.
C.elegans : une plateforme novatrice
Ii) ÉTABLISSEMENT D’UN MODELE DE RETARD MENTAL CHEZ
C.elegans.
III)
IDENTIFICATION DES CIBLES THÉRAPEUTIQUES CONTRE
CERTAINES Formes de retard mental
1) La génération et la prédiction des
interactions génétiques chez C.elégans
· Le modèle statistique
· Résultat
2) Discussion
IV) CONCLUSION
V) Bibliographie
I) Introduction
1) Biologie des
systèmes et génomique fonctionnelle :
La biologie des systèmes (génomique intégrative) à pour
but d'étudier le fonctionnement d'un organisme vivant (un fragment de tissu, cellule, réseaux de gènes et de protéines permettant la
communication des cellules) sont des
systèmes complexes. Ces systèmes regroupent de nombreuses fonctions
physiologiques qui interagissent entre elles. Les techniques issues de la
génomique permettent de recueillir des données biologiques produites par les
systèmes. Ces données, fort nombreuses, doivent être intégrées avant leurs
interprétations.
La génomique a pour objectifs d'analyser la
structure du génome, de séquencer les gènes et d’identifier leurs
fonctions. La génomique fonctionnelle étudie la régulation de
l'expression des gènes à l'échelle d'une cellule, d''un organe ou d'un
individu. On peut analyser l'expression du génome au cours du temps (pendant le
développement embryonnaire) ou comparer deux états (sain vs.malade). Une grande
quantité et hétérogénéité caractérisent les données issues de la génomique
fonctionnelle. L'exploitation efficace de ses données repose sur
l'ensemble des approches mathématiques, algorithmiques et statistiques
appliquées à la génomique. Elles permettent l'intégration des données obtenues
par diverses technologies. Il est même possible, grâce à des systèmes de
gestion de l'information, de compiler les résultats obtenus dans les
plateformes de biologie intégrative et de les comparer avec la littérature
scientifique préexistante. Aujourd'hui, certains logiciels sont capables de
fouiller la littérature scientifique à la recherche de paramètres cliniques
mesurables ou non.
La combinaison des données hétérogènes produites par la génomique
fonctionnelle dans de gigantesques bases de données, pour aussi efficace
qu'elle soit, ne peut suffire à comprendre les mécanismes biologiques. En
effet, une fonction biologique ne résulte pas de la simple addition des
propriétés de ses composants élémentaires (ARN, protéines...) mais des propriétés
induites par leurs nombreuses interactions. C'est pourquoi il faut ajouter le
problème attaché à l'analyse des réseaux d'interactions complexes. Il est donc
nécessaire d'adopter une démarche (nouvelle en biologie) qui s'appuie sur des
concepts issus des mathématiques appliquées, et plus spécifiquement la théorie
des systèmes complexes.
2) Démarche suivie dans
la biologie des systèmes :
La biologie des systèmes adopte une démarche itérative et
intégrative en combinant des approches expérimentales et théoriques dans
les quelles les mathématiques jouent un rôle central.
Dans un premier temps, l'hypothèse de départ est formulée
à partir des connaissances scientifiques disponibles sur un système à un moment
donné. Ces connaissances sont sous la forme de données produites par un ou
plusieurs niveaux hiérarchiques de l'organisation des systèmes biologiques.
Elles seront intégrées dans un modèle mathématique préliminaire afin d'établir
des prédictions sur le fonctionnement de ce système biologique.
Dans un deuxième temps, certains éléments du système
seront perturbés expérimentalement. On compare l'état perturbé à un témoin
normal (modèle expérimental de maladie, souris knock out, knock in, gene
silencing et l'interférence d'ARN (RNAi). Les résultats obtenus ne seront pas tous
conformes au modèle mathématique de départ. Ce qui conduit à modifier les
hypothèses initiales.
Dans un troisième temps, les hypothèses de travail
modifiées serviront à concevoir en retour d'autres perturbations expérimentales
du système. Ce raisonnement sera répété autant que nécessaire jusqu'à ce que
les résultats expérimentaux se superposent avec le modèle mathématique. (Figure
1)
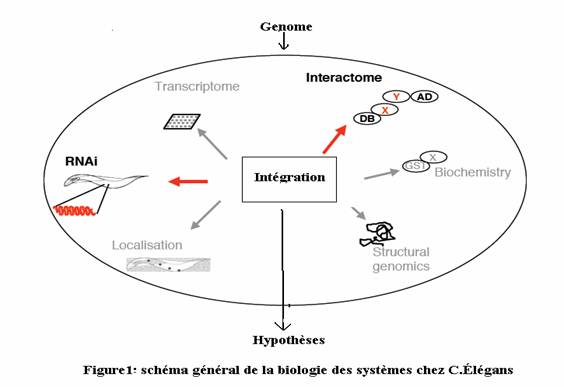
Figure1 : schéma général
de la biologie des systèmes chez C.élégans
3) Les petites
protéines G :
Les protéines G, dites G car elles
fixent un nucléotide guanylique (GTP ou GDP), sont douées d’une activité GTPase
intrinsèque. On oppose par leurs tailles et leurs fonctions :
Les grandes protéines G, hétérotrimétrique
(formées de 3 sous-unités alpha, béta, gamma), qui sont associées aux
récepteurs couplés aux protéines G.
Et les petites protéines G, monomériques
ancrées de façon covalente à la face interne de la membrane plasmique, qui sont
impliquées dans diverses voies de signalisation
Les petites protéines G composent la super
famille de Ras très conservée entre les espèces. Cette super famille comprend 5
sous-groupes : Ras, Rho, Ran, Rab et le sous famille des Arf. Chez
l’homme, environ 150 de ces protéines de signalisation sont exprimées et
servent à réguler la croissance, la mobilité cellulaire, la morphogenèse et le
trafic membranaire. L’activation de ces petites protéines G, qui passent d’un
état inactif liées au GDP à un état actif liées au GTP est facilitée par des
protéines régulatrices, en particulier les facteurs d’échange nucléotidiques
GEFs (Figure 2). D’autres protéines régulatrices sont associées aux
petites protéines G :
- Les protéines GDS (Guanine
nucleotide Dissociation Stimulators), comme p. ex. la protéine adaptatrice
Sos.
- Les protéines GDI (Guanine
nucleotide Dissociation Inhibitors),
- les protéines GAP
(GTPase-Activating Proteins) qui activent l’activité GTPase(2)
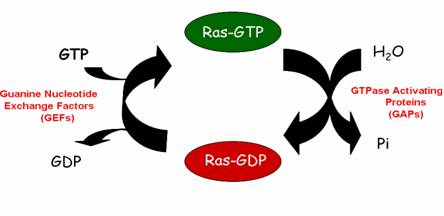
Figure 2 :
Les Cycles d’échange GTP/GDP de Ras
Ras est une molécule intégratrice, un seul signal
d’activation peut induire plusieurs effecteurs, ces petites protéines G peuvent être
considérées comme des interrupteurs moléculaires des voies de
signalisation : en position « ON », c'est-à-dire liées au GTP : Le
signal passe, en position « OFF », c'est-à-dire liées au GDP: Le signal ne
passe pas (Figure 3) (2)
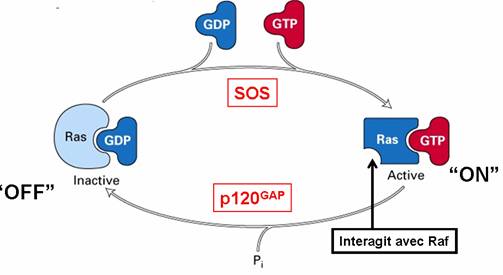
Figure 3 :
Fonction des petites protéines Ras comme des interrupteurs moléculaires
des voies de signalisation
La perturbation de la régulation des protéines Ras est à
l'origine de 17 pathologies chez l'humain, dont le développement des cancers et
de certaines pathologies reliées au retard mental (Figure 4) (3)
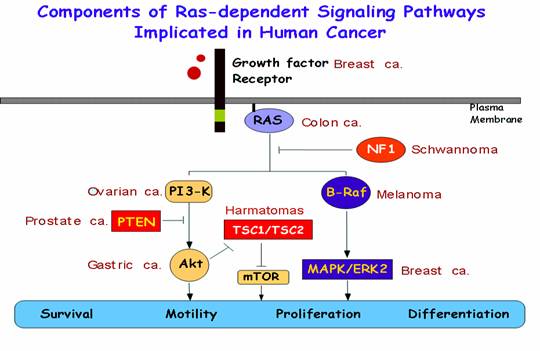
Figure 4 :
Schéma de l’implication des composés de la voie de signalisation de Ras dans
les différents types de cancer
4) C.elegans : une plateforme
novatrice
Dans le but d’étudier les mécanismes de régulation des
petites protéines G : leurs compositions, connectivités et
l’identification de cibles thérapeutiques pour les 17 pathologies
impliquant les GTPase Ras, un modèle animal à été adapté aux études de
génétiques a haut débit : C. elegans, il s’agit de l’animal le plus connu
sur terre car la localisation et la lignée de chaque cellule de l’embryon, de
la larve et de l’adulte sont connues. C'est un organisme pluricellulaire
simple, suffisamment proche des organismes pluricellulaires supérieurs, ce qui
en fait un très bon modèle pour l'étude du développement embryonnaire et de la
signalisation cellulaire. De plus, son génome a été le premier génome animal à
être complètement séquencé : il contient 19099 gènes, dont 200 peuvent
être rendus létaux par mutation. La carte génomique rend l’identification d’un
gène dont lequel se trouve une nouvelle mutation très facile. Le criblage
génétique est facile parce qu’il est possible d’examiner un grand nombre de
vers, et le temps de génération n’est que 3 jours. Ce sont des hermaphrodites
autofécondés. Il est facile de fabriquer des vers transgéniques par
injection de l’ADN nécessaire dans la gonade. Comme il est transparent, des
vers transgéniques exprimant des protéines fluorescentes
vont permettre l’identification d’un grand nombre de
mécanismes moléculaires et cellulaires sur l'animal vivant par microscopie à
fluorescence. Il est aussi facile d’inhiber l’action génétique maternelle par
injection d’ARN double brin complémentaire de l’ARN messager cible dans les
gonades, ce qui conduit a la destruction de l’ARNm spécifique par un mécanisme
catalytique (1).
Chez les organismes multicellulaires, le développement
embryonnaire et la survie cellulaire sont dépendants d'un flot continuel
d'informations transmises au sein d'un réseau complexe de cellules. Afin
d'adopter un comportement approprié à leur environnement, les cellules ont
besoin de capter, d'intégrer et d'interpréter ces informations. Les machineries
moléculaires responsables de l'intégration de ces différentes informations sont
encore peu connues. Avec ce modèle on peut mieux comprendre le rôle joué par
certaines de ces machineries à différentes étapes du développement et de la vie
du ver : les machineries de régulation et de la coordination des GTPases de la
superfamille des Ras. C. elegans, contient 150 gènes, elles coordonnent
leurs actions dans différents types cellulaires et à différentes étapes du
développement et de la vie du ver afin d’obtenir une vision globale de ce
système et de sa dynamique dans le temps et l'espace (Figure 5) (1)

Figure 5 : C.elegans un modèle animal adapté aux
études de Génétiques à haut débit.
II) Établissement d’un modèle de retard mental
chez C.elegans :
Des expériences d'interférences d'ARN (RNAi) (4) ont
permis d’analyser de façon systématique la fonction des gènes de
Caernorhabditis elegans. Il a été montré que les fonctions physiologiques dont
l’apprentissage, mémorisation, la morphogenèse, l’innervation du pharynx et le
contrôle de la fréquence de reculon en phase de recherche de nourriture sont
régulées par de nombreuses voies de signalisation qui interviennent dans la
plasticité synaptique (5) et impliquant plusieurs protéines (tels que des
récepteurs membranaires, des petites protéines G comme Ras).
Ces expériences ont montré le rôle de gdi-1
dans la fertilité du ver, et plus précisément dans la morphogenèse des gonades.
En fait, gdi-1 associer avec la protéine G de la famille Ras, rab-5 sont
requises dans la maturation et/ ou ovulation des oocytes.
III) Identification
des cibles thérapeutiques contre certaines formes de retard mental :
La plateforme C. elegans est utilisée dans l'identification de cibles thérapeutiques contre certaines formes de retard mental. L’hypothèse de départ, c’est qu'il y a un déséquilibre dans la machinerie de régulation des GTPases de la superfamille des Ras chez l'humain, ce qui se traduirait par une perturbation du fonctionnement de neurones impliqués dans les processus de mémorisation. Ce déséquilibre pourrait être réversible ; on pourrait donc en théorie rééquilibrer la machinerie de signalisation déficiente par voie chimique et ainsi obtenir une réduction des symptômes par une interaction tamponnante. (Figure 6A). Les cibles thérapeutiques peuvent être soit des récepteurs membranaires ou des protéines entrant dans la cascade de la vois de signalisation incluant les GTPase Ras . (Figure 6B)
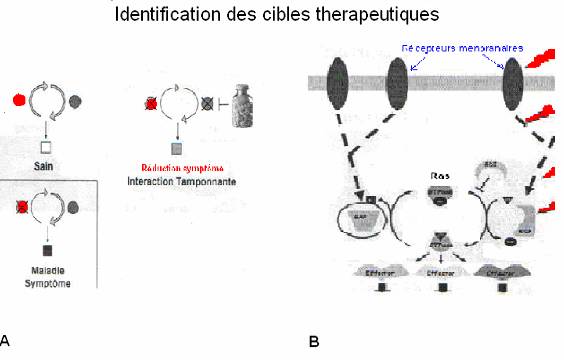
Figure 6 : A) un équilibre entre deux effets chez une personne saine,
chez une malade, on a un déséquilibre
par inhibition d’un des deux effets. Le
traitement chimique réduit les symptômes avec une interaction tamponnate. B)
représente les cible thérapeutiques éventuelles dans la vois de
signalisation des MAPK ase.
1) La génération et la prédiction des interactions génétiques
chez C.elégans (Weiwei Zhong et al.
2006) :
Pour prédire les interactions génétiques du grand génome C.elégans et obtenir une vue globale des interactions fonctionnelles parmi les gènes dans le génome du métazoaire, ces interactions incluent les interactions physiques protéine-protéine comme bien gène-gène et protéine gène. Nous intégrons statistiquement les données du double hybride, données des expressions des gènes, données phénotypiques et les données d’annotations fonctionnelles à partir de trois organismes modèles S.C et C.elegans et DM. Puisque Seulement 292 gènes C.elegans ont une complète annotation de l’expression anatomique, phénotypique et processus biologique dans Gene Ontology (GO), sachant que les fonctions des gènes sont souvent conservées au niveau moléculaire et que si deux gènes possèdent des faits indiquant une interaction génétique, leurs gènes orthologues ont aussi la même liaison fonctionnelle. La quantité d’informations à travers les espèces peut détecter les interactions même si les données génétiques d’un organisme sont incomplètes.
L’ensemble des données à partir des différentes sources peut avoir un taux d’erreur intrinsèque, ce qui nécessite un modèle statistique solide pour produire une bonne prédiction (6,7)
- Le modèle statistique
Pour intégrer
les données biologiques, la méthode utilisée est la régression logistique
(8) qui est une
technique statistique qui a pour objectif, à partir d’un fichier
d’observations, de produire un modèle permettant de prédire les valeurs prises
par une variable catégorielle, le plus souvent binaire (interaction vs pas
d’interaction), qui donne des résultats comparables à la méthode naïve Bayesian network.
Le calibrage des paramètres du système
informatique se fait par la construction du set training pour les
interactions génétique de C.elegans. Les positifs sont représentés par
1816 paires d’interactions génétiques provenant de la littérature et 2878 paires d’interactions physiques
identifiés par double hybrides.
Les négatifs sont représentés par les protéines avec des localisations
subcellulaires différentes pour les interactions physiques. Les négatives de
l’interaction génétique représentent les doubles mutants des gènes existant et qu’aucune interaction est rapportée, ce qui représente
3296 paires d’interactions prisent à partir de la base de données WormBase
(9).
L’algorithme de prédiction des interactions commence par la cartographie des gênes orthologues en utilisant le InParanoid (Un serveur InParanoid (Remm et al. 2001) pour la recherche d’orthologues entre différentes espèces). La sélection des paires de gènes de C.elegans orthologues aux paires dans D.melanogaster et S.cerevisiae se fait pour 5 faits :
1. L’expression
anatomique identique
2. Phénotype
3. Annotation
fonctionnelle
4. Coexpression
microarray
5. Présence des
interlogues.
Le ratio de likelihood
assigne un score pour chaque caractéristique (feature), il est défini comme la
fréquence de l’ensemble des positifs par apport à la fréquence du jeu de donné
négatif :
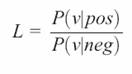
Si le ratio likelihoode est 1, il n’y a pas de différence
entre les données.
Sinon si le ratio likelihoode est > 1 : indique
des prédicateurs positifs, c a d que le feature est fortement présent dans les
interactions des paires de gène. Les paires de gène avec un haut score
indiquant une forte prédiction
Sinon si le ratio likelihoode est < 1 : indique
des prédicateurs négatifs
- Résultat :
Les paires de gène
avec un fort coefficient de corrélation person (10) dans les résultats
de microarray obtient un haut L-score que les paires avec un faible
coefficient.
Les paires de gène avec une expression spécifique de
phénotype ou annotation fonctionnelle donne un haut L-score que les paires de
gène avec une annotation générale ou pas d’annotation.
Les scores sont intégrés pour estimer les probabilités
globales d’interaction entre deux gènes de C.elegans. (Figure7)
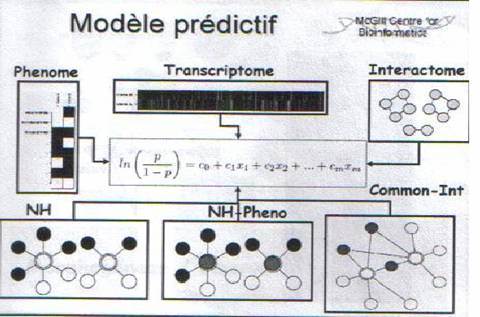
Figure 7 : Le calcule de la probabilité globale d’interaction entre les
gènes de C.elegans à partir des données
biologiques fournies des interactomes, phenome et transcriptosome. Le p
est le score output final qui vari entre 0 et 1 où 1 indique la paire avec une
interaction génétique et 0 : pas d’interaction.
En appliquant un seuil de 0.9 qui représente la contribution maximale que chaque feature unique puisse achever, on obtient le résultat, qui un Network d’interaction génétique pour 2254 gènes et 18183 interactions qui fourni un framework de la compréhension des fonctions du gène
Les gènes sont regroupés en se basant sur leurs interactions avec leurs partenaires.
Les regroupements révèlent une organisation modulaire du network. Plusieurs groupements sont corrélés avec un complexe protéique ou un processus biologique (Figue 8) et (Figure 9)
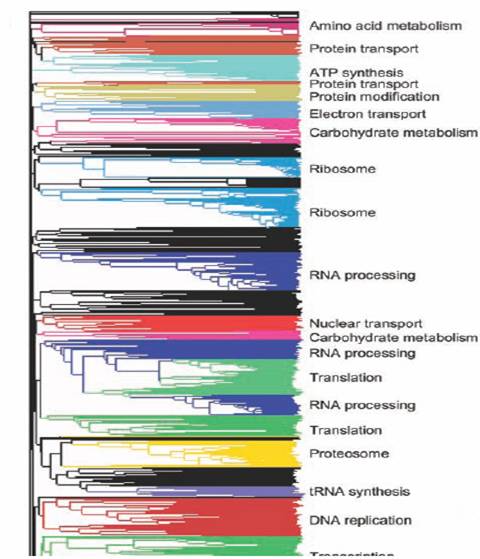
Figure
8 : Représente une partie de
l’arbre des regroupements de gènes, en se basant sur leurs distances dans
network de prédiction. Le cluster est coloré et annoté si le groupement de
gènes montre une fonction commune. Les clusters avec une fonction inconnue sont
en noire
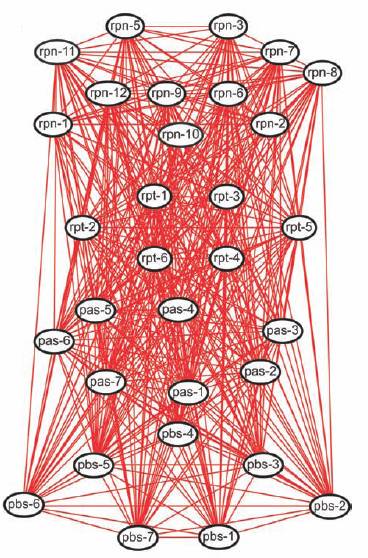
Figure 9 : Une vue locale d’un module du Network,
le regroupement des protéines du protéasome
Le système prédit plusieurs interactions dans la voie de signalisation des MAPKs, où on trouve le gène let-60 impliqué dans le développement de la vulve et qui code pour la protéine Ras chez C.elegans (11) (Figure 10).
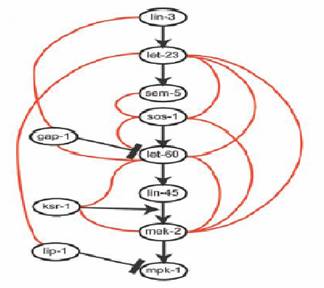
Figure 9 : Une représentation locale de la
voie de signalisation impliquant
EGF-ras-MAPK. Les flèches sont des activateurs et les bars sont des
inhibiteurs
2) Discussion :
La voie de signalisation des MAPKase impliquant Ras est requise dans la plasticité synaptique. Gdi-1 possède une activité inhibitrice sur l’activité catalytique des GTPase de la famille des Ras, plus précisément sur la protéine Rab et pourra être une cible thérapeutique dans certaines maladies génétiques humaines (comme le retard mental).
Le modèle statistique prédit 70 interactions pour gdi-1. Des analyses épistasiques (12) permet de déterminer parmi ces gènes prédits, lesquelles induisent la suppression du phénotype dans la voie métabolique.
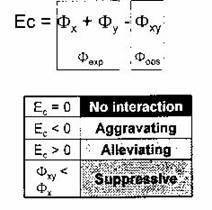
Plusieurs expériences (Sarah Jenna et al. 2007) montrent
que parmi les gènes prédits, ceux qui ont une interaction génétique avec gdi-1
et qui régulent son activité avec une bonne spécificité sont les gènes codant
pour les protéines DYB-1 et Crb2. Ces dernières se lient à un récepteur situé sur la membrane plasmique
(DGC) Dystrophin-glycoprotein complex des
cellules nerveuses. Probablement, le récepteur DGC pourrait interagir avec le
récepteur AMPAR (13) au cour de la plasticité synaptique.
Une altération au niveau du récepteur DCG, ainsi que dans
la voie de signalisation impliquant Ras et Rab pourrait être impliquée dans le
retard mental. (Priya
Sethu Chockalingam et al. 2002).
Dans la prédiction des interactions génétiques dans
C.elegans. Un certains nombres de gènes forment un cycle de voisin dans le
network. Il s’agit des gènes pix-1, git-1 et pak-1. Ces gènes jouent un rôle
dans la régulation de la morphogenèse et l’innervation du pharynx du ver.
IV) Conclusion :
L’approche de la génomique intégrative permet
l’identification des cibles thérapeutiques contre des formes de retard mental
liées à l’altération de la machinerie de la régulation des GTPase Ras. Ces études sont réalisées sur le nématode C.elegans, qui
représente une plateforme novatrice pour réaliser la prédiction des gènes
potentiels dans l’évolution de certaines maladies génétiques.
V) Bibliographies :
1) Livre biologie du développement de J.M.W. Slack
2) Livre Biologie moléculaire. Biochimie des communications
cellulaires de Christian Moussard
3) Bos JL. ras oncogenes in human cancer: a
review Cancer Res., 49(17):4682-9. 1999
4) http://en.wikipedia.org/wiki/RNA_interference
5)
http://fr.wikipedia.org/wiki/Plasticit%C3%A9_synaptique
6)Weiwei Zhong and Paul W. Sternberg. Automated data
integration for developmental biological research. Development, 134(18): 3227-3238.
(2007)
7) Weiwei Zhong and Paul W. Sternberg.
Genome-wide
prediction of C. elegans genetic interactions. Science, 311(5766): 1481-1484. (2006)
8) P.M.Preux and P.Odermatt Q’est ce qu’une régression logistique? Rev Mal Respir : 22 : 159-160 62 (2005)
9) Base de donnée du nématode : www.wormBase.com
10) http://www.u707.jussieu.fr/biostatgv/pearson.php