De la biologie intégrative à l’identification
de nouvelles cibles thérapeutique
Par Hocine AB
Plan
c/
Données de régulation de la transcription.
e/
Ontologies et annotations fonctionnelles
III
Comment intégrer les données biologiques
b/
Fusion des 3 profils d’interaction
c/
Prédiction de la fonction de gènes
V
Application dans le monde de l’industrie
Ce rapport a été
réalisé dans le cadre du cours BIF7002 donné par le professeur Vladimir Makarenkov à l’Université du
Québec à Montréal.
Ce rapport est destiné
à un public scientifique et a pour but de sensibiliser ce public au domaine de
la biologie intégrative computationnelle.
Cette étude s’appuie
sur les résultats de différentes publications scientifiques dont vous trouverez
les références à la fin du rapport.
I Introduction
Dans un premier temps,
il est important de définir ce que l’on entend par « biologie
intégrative ».
En effet, cette
expression peut être entendue dans différents sens, et notamment dans le sens
de biologie systémique. Or, ce n’est pas l’acception que nous utiliserons ici,
c’est pourquoi nous ajouterons le qualificatif « computationnelle »
pour marquer la différence.
La biologie intégrative computationnelle
est l’intégration d’une masse importante de données hétérogènes, moléculaires (génomique,
transcriptomique, protéomique,
métabolomique) ou autres via des outils informatiques
et mathématiques. Les données à intégrer peuvent concerner différentes espèces.
La finalité principale
de l’utilisation de la biologie intégrative computationnelle est de pouvoir
exploiter de la façon la plus efficace possible l’ensemble des données biologiques
possibles.
Nous allons dans cette
étude nous intéresser à l’intérêt et aux limites de la biologie intégrative
computationnelle et à ses applications dans le domaine de la recherche
fondamentale, mais aussi à ses applications dans l’industrie.
II Les types
de données
a/ Les
données d’expression
Ces données
correspondent à ce qu’on appelle le transcriptome. Le
transcriptome est l'ensemble des ARNm issus de l'expression d'une partie du génome d'un
tissu cellulaire ou d'un type de cellule.
Ce type de données est
généralement obtenu de 2 manières :
-
des
expériences in vivo comme par exemple l’hybridation in situ, qui offrent une
bonne résolution spatiale et temporelle, mais qui ont pour inconvénient le
bruit inhérent à l’expérience.
-
Les puces
à ADN, qui souffrent du manque de standardisation entre les différentes
plateformes, notamment au niveau de la présentation des résultats.
b/ Interactome
L’interactome
est l’ensemble des données sur les interactions physiques entre les molécules
biologiques d’une cellule, il peut s’agir d’interactions protéine-protéine mais
aussi d’interactions entre gènes.
Une des principales
techniques utilisées est Y2H (Yeast two-hybrid assay)
ou double-hybride. Des expériences Y2H ont été menées sur l’intégralité des
génomes de l’homme, du ver et de la mouche. Donc, on dispose d’une quantité
importante d’informations sur les interactions protéine-protéine. En
comparaison, peu de données sont disponibles sur les interactions génétiques.
La technique Y2H présente
un haut taux de faux positifs : en effet, il peut exister des interactions
entre des protéines, alors qu’in vivo elles ne sont jamais synthétisées au même
endroit ou au même moment. Il est donc nécessaire de recouper les résultats
obtenus.
c/ Données
de régulation de la transcription.
Les données sont
principalement obtenues par la méthode ChIP (chromatin ImmunoPrecipitation)
qui détecte les interactions protéine-ADN in vivo. Cette technique a ses faiblesses puisqu’une protéine peut se lier à une
séquence d’ADN sans qu’il y ait de conséquence, on observe du coup un certain
taux de faux positifs.
d/ Les
données phénotypiques
Les données
phénotypiques d’un gène portent sur les conséquences produites par l’altération
de ce gène. Ces données sont ajourd’hui
essentiellement obtenues par :
-
ARN
interférent
-
Knock Out
ciblé
e/ Ontologies
et annotations fonctionnelles
Dans le sens général,
l’ontologie est l'ensemble structuré des termes et concepts fondant le sens
d'un champ d'informations, et son objectif premier est de modéliser l’ensemble
des connaissances dans un modèle donné.
Donc, le but est ici
de limiter la confusion des voix autour de l’annotation de l’information
moléculaire sur les gèns et leur produit.
Par exemple, une gène ayant l’annotation « létalité embryonique » ne serait pas, sans l’utilisation d’une
ontologie phénotypique, associé fonctionnellement à un gène annoté
« défaut de gastrulation », alors que le défaut de gastrulation mène
à une létalité embryonnaire. C’est cette perte d’information que l’ontologie
est censé éviter.
Le
GO (Gene Ontology) Consortium (2006) fournit une des
plateformes d’annotations fonctionnelles de gènes les plus utilisées. Ce travail a donné naissance à trois
ontologies respectivement consacrées aux fonctions
moléculaires, aux processus
biologiques et aux composants
cellulaires. Il est à noter que Gene Ontology est
de plus en plus utilisé par la communauté des biologistes. Ceci s’explique
notamment par le fait que les
données de Go sont contrôlées à la main.
De par sa qualité, GO
est non seulement utilisé comme un ensemble de données potentiellement
intégrables, mais aussi comme un moyen de contrôler les performances de
méthodes d’intégration de données. Sa seule limite est le manque d’informations :
en effet, de nombreux gènes ne sont associés à aucune annotation, Chez C.elegans, 80% des gènes ne sont pas annotés. Une solution
possible pour remédier à ce manque d’informations, est de croiser les données
de plusieurs espèces, donc en prenant compte des gènes orthologues.
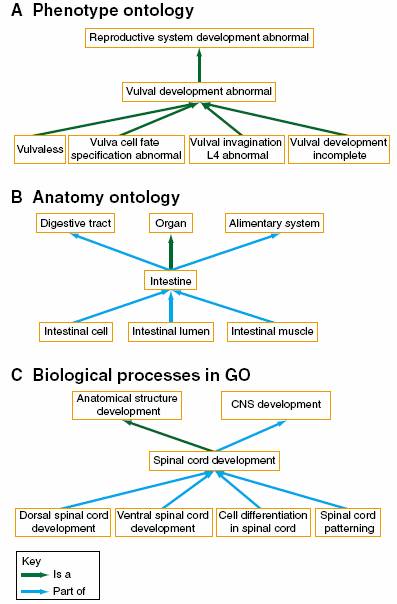
Exemples de bio-ontologies
(source GO)
f/ Conclusion
L’utilisation combinée
de l’ensemble de ces données a pour but de limiter le champ d’investigation,
mais pour cela les données utilisées doivent être fiables et donc de très bonne
qualité.
Une autre limite est
le manque d’informations, en effet l’intégration de données ne remplace pas le
travail du biologiste dans le screening du génome, mais le valorise; le
biologiste doit donc continuer à produire des données. Et son rôle est aussi
important dans la validation des conclusions tirées de l’intégration de
données.
III Comment
intégrer les données biologiques
L’intégration se fait
par des traitements statistiques à travers des outils informatiques. Donc, une
des premières tâches est de traduire ces informations dans un langage compris
par l’ordinateur, une manière de le faire est de par exemple convertir en code
binaire la présence ou l’absence d’une annotation pour chacun des gènes d’un
génome. Mais, il faut aussi être vigilant à ne pas perdre de l’information au moment
de l’encodage des données dans un format compréhensible par un ordinateur, donc
quand ce sera possible, il faudra pondérer ou scorer ces données, pour par
exemple préciser la fiabilité des données en question.
Le traitement des
données pose aussi la question du choix du modèle statistique à utiliser. Le
modèle le plus simple est le système de
vote : il suffit de choisir le nombre de fois qu’une interaction doit
être vérifiée pour être retenue; ce nombre varie de 1 à n, n étant le nombre de
types de données utilisés (interactome, transcriptome...).
Des modèles
statistiques plus sophistiqués peuvent être utilisés comme les arbres de
décisions, les réseaux bayesiens ou la régression
linéaire, mais pour cela il faut disposer de « training » sets.
Mais, il s’avère
finalement que le choix du modèle statistique n’a que peu d’incidence sur les
résultats obtenus. Le goulot d’étranglement reste l’étape de collection de
données, notamment sa qualité et sa quantité.
IV Application
pratique
Dans un article publié
dans Nature (cf ref. 2),
Gunsalus et ses collaborateurs nous font la
démonstration de l’efficacité et de la puissance de la biologie intégrative
dans l’étude des machines moléculaires impliquées dans l’embryogénèse précoce
de C. elegans.
Leur expérience a lieu
en 2 étapes :
-
La 1ère
étape consiste à intégrer toutes les données possibles concernant le modèle
choisi, soit : des données sur le profil phénotypique, l’interactome, et des données concernant le profil
d’expression.
Et à partir de ces données, ils ont généré un réseau d’interactions
fonctionnelles durant l’embryogénèse précoce chez C. Elegans
-
La seconde
étape consiste à utiliser le réseau d’interactions généré pour prédire le rôle
et l’implication de certains gènes et protéines de C.elegans
durant l’embryogénèse précoce.
a/ Intégration
des données
Le but de cette étape
est de bâtir des modèles de machines moléculaires impliquées dans
l’embryogénèse précoce, et des connexions fonctionnelles entre ces machines. De
tels modèles peuvent être déduits d’un réseau gène-protéine, lui-même généré
par 3 types de de relations fonctionnelles : les
interactions protéines-protéines (interactome), une
carte des profils d’expression de gènes (transcriptome),
et un profilage phénotypique des gènes.
L’intégration de
l’ensemble de ces 3 types de données est réalisé en
concevant un graphe dont chaque noeud correspond à un
gène ou au produit de ce gène (ARN ou protéine), et chaque arête correspond à
une relation fonctionnelle d’un des 3 types décrits ci. Les régions hautement interconnectée
représentent chacune une machine moléculaire ou un processus biologique.
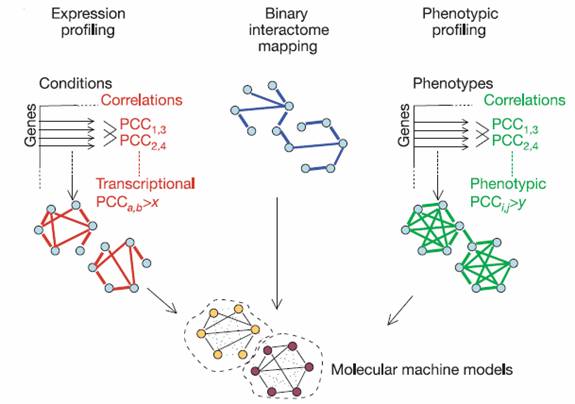
Modèle d’intégration des données dans le but de modéliser machines
moléculaires (Source Gunsalus et al. (2005) )
1- Interactome utilisé
L’équipe de Gunsalus a utilisé un interactome
(W17) généré à partir des résultats d’autres équipes, c’est-à-dire une
combinant un interactome (W15) avec un set d’ « interologs » fly-to-worms, ces interologues sont des
protéines de D.melanogaster qui interagissent entre
elles et qui possèedent des homologues chez C.elegans. Il a ainsi obtenu un interactome
de 3848 protéines liées par 6572
interactions.
Gunsalus a évalué la cohérence de l’interactome
obtenu en comparant les interactions entre les protéines synthétisées lors de
l’embryogénèse précoce, et celles de protéines prises au hasard dans le protéome de C.elegans. Sans
surprise, les protéines exprimées lors de l’embryogenèse montre
une plus grande connectivité.
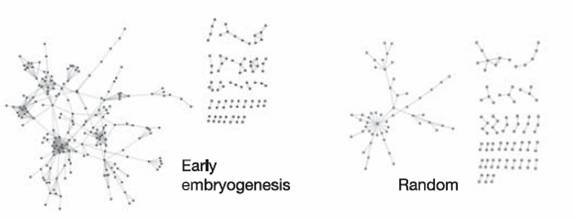
Le
sous-réseau représente les protéines exprimées lors de l’embryogenèse. Celui de
gauche représente des protéines prises au hasard. Le sous-réseau de gauche
montre une plus grande connectivité.
2- Transcriptome
utilisé
Pour
le transcriptome, Gunsalus
utilise aussi les résultats d’une autre équipe obtenus par des expériences de
puces à ADN, mais il applique à ces résultats un certain seuil par la méthode
PCC (Pearson correlation coefficient).
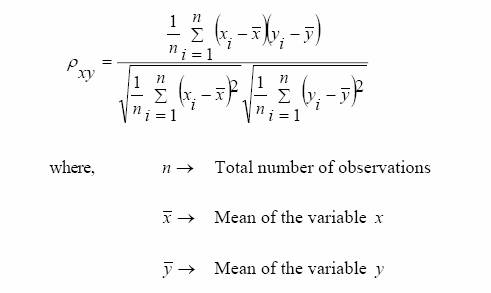
PCC mesure le degré d’association entre «
variables »
3- Profilage
phénotypique
Le clustering par similarité phénotypique a été obtenu en
appliquant un seuil sur le coefficient de corrélation (PCC) calculé sur les
résultats d’expérience ARN interférent sur les 661 gènes identifiés de l’embryogénèse
précoce.
Gunsalus a
identifié 23 phénoclusters.
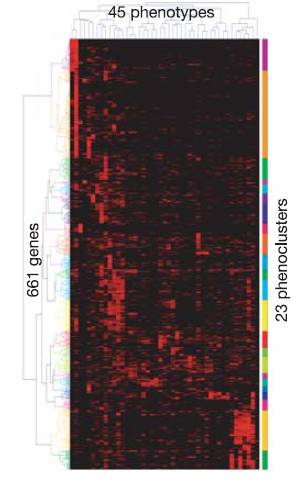
Clustering par similarité phnénotypique des 661 gèenes de
l’embrypogenèes précoce chez C.elegans.
(Source Gunsalus et al. (2005))
Gunsalus constate une forte corrélation entre le niveau
de similarité phénotypique entre 2 gènes et le fait qu’ils partagent des attributs
fonctionnels similaires (GO).
Cela confirme
l’intérêt de la méthode.
b/ Fusion
des 3 profils d’interaction
Afin d’évaluer la
pertinence de fusionner les résultats obtenus par les 3 types de données (interactome, transcriptome,
données phénotypiques), Gunsalus a mesuré le niveau
de corrélation entre les résultats obtenus. Il a pu observé
un haut niveau de corrélation.
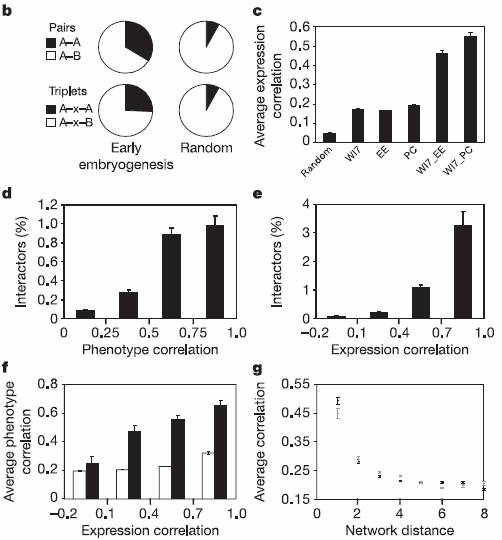
Corrélations
entre les différents set de données (Source Gunsalus et al. (2005) )
Gunsalus a donc pu procédé à la fusion des données
obtenues, et il a ainsi créé un réseau (graphe) de 661 gènes/protéines
(sommets) et 31173 interactions (arêtes).
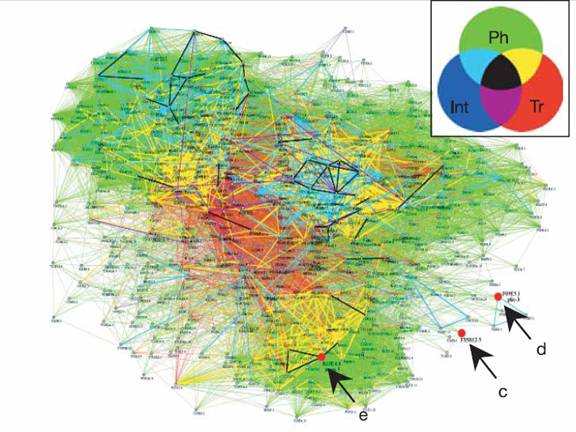
Réseau d’intégration de tous les sets de données. Le diagramme de venn (en haut à gauche) correspond aux couleurs des arêtes
en fonction des sets de données qui supportent les interactions représentées
par l’arête. (Source Gunsalus et al. (2005) )
Mais Gunsalus est allé plus loin, puisque il a pris en compte le
type d’interaction qui lie chaque paire de gènes/protéines : « Int »
pour les interactions protéines/protéines, « Tr » pour les
similarités d’expression, et « Ph » pour les similarité phénotypique,
pour ensuite, à partir du graphe décrit ci-dessus, retenir un-sous-graphe ne
contenant plus que les arêtes correspondant à au moins 2 des 3 types d’interactions Int, Tr
et Ph. Il a ainsi obtenu ce qu’il appelle « le réseau à multiple
support », qui ne contient plus que 305 des 661 noeuds
initiaux(gènes/protéines); ce nouveau graphe révèle
des groupes distinct de noeuds fortement
interconnectés, et avec peu d’interactions entre les groupes.
Gunsalus a identifié 2 types de groupes :
-
les
groupes dont les noeuds sont interconnectés par des
liens Int et Ph (ou par les 3 types).
Ces groupes correspondent à des complexes moléculaires connus. Et grâce
aux annotations fonctionnels (GO), Gunsalus a pu
identifier chacune de ces machineries, dont entre autres : ribosome, proteasome, APC (anaphase-promoting
complex) .
-
les
groupes dont les arêtes portent une corrélation Ph et Tr. Ces groupes
correspondent à des gènes impliqués dans des processus cellulaires fonctionnels
interdépendants : comme par exemple, le métabolisme ARNm/protéine
(transcription, contrôle de la traduction, modification et trafic des
protéines)
Ainsi, pour chacun des
groupes, en se basant sur les annotations fonctionnelles, Gunsalus
va associer soit une machinerie moléculaire, soit un ensemble de processus
cellulaires interdépendants.
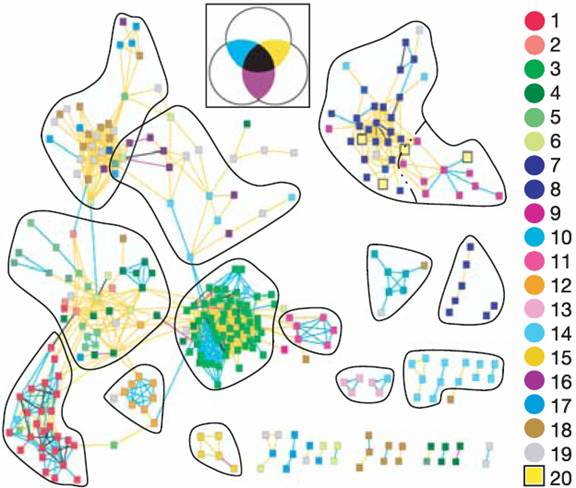
Multiple support Network ou réseau à support multiple (Source Gunsalus et al. (2005) )
c/ Prédiction
de la fonction de gènes
À partir du «
réseau à support multiple », Gunsalus a
sélectionné 10 gènes dont la fonction est inconnue, mais associés à un des
groupes de gènes du réseau, donc à une machinerie moléculaire; le but étant
bien-sûr de vérifier si les gènes en question sont bien impliqués dans la
machinerie moléculaire à laquelle le graphe les associe.
Pour cela, Gunsalus a utilisé une méthode différente de toutes celles
utilisées pour générer l’ensemble des données : utilisation de la protéine
fluorescente verte (GFP, cf Références) suivie par l’observation in vivo par
microscope à fluorescence. La localisation au sein de la cellule et en fonction
du temps de la protéine issue du gène d’intérêt pourra confirmer ou infirmer la
participation du gène/protéine à la machine moléculaire prédite.
Par exemple, l’un des
10 gènes sélectionnés est supposément impliqué dans la fonction centrosomale, et son observation par microscopie à
fluorescence a démontré une présence dans 2 zones périnucléaires
distinctes (présence compatible avec celles de centromères), et un comportement
dynamique (déplacement) tout à fait conforme à celui des centrioles. La
prédiction a donc été confirmée par l’expérience d’observation in vivo.
Pour 8 des 10 gènes
sélectionnés, la prédiction a été vérifiée de façon évidente; pour les 2
autres, la prédiction n’a pu être infirmée ni confirmée.
Il est inutile d’appliquer un traitement statistique à ces
résultats pour montrer qu’ils n’ont pas été obtenus par hasard. Gunsalus a démontré que l’étude du profilage phénotypique
combiné à l’interactome et au transcriptome.
Il a prouvé ainsi que
la biologie intégrative computationnelle est un outil puissant de prédiction
fonctionnelle, et cet outil peut être utilisé dans un cadre bien plus large que
l’embryogénèse précoce chez C. elegans.
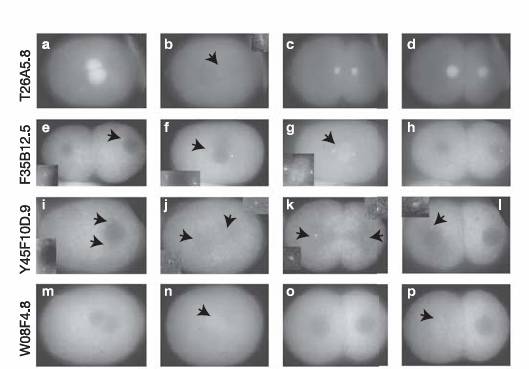
Pattern de localisation embryonique de
protéines fusionnées à GFP qui confirme le modèle de prédiction (Source Gunsalus et al. (2005) )
V Application
dans le monde de l’industrie
Les cibles
thérapeutiques sont les molécules de l’organisme sur lesquelles les médicaments
développés par l’industrie agissent. La recherche de cible a lieu en 3
étapes :
-
la
découverte, que l’on peut apparenter à la création du réseau à support multiple
présenté dans le chapitre précédent
-
la
sélection, que l’on peut comparer à la sélection des 10 gènes inconnus par Gunsalus
-
la
validation, que l’on peut comparer à la validation opérée par Gunsalus par l’expérience de microscopie à fluorescence
Le monde de la
découverte et de la validation de cibles thérapeutiques est émergent et très
prometteur, car en agissant très en amont dans la chaîne de développement des
médicaments, on peut diminuer considérablement le coût de développement de
médicaments.
En général, le taux de
validation des cibles sélectionnées est de 5 à 10%. Mais, une entreprise comme Helios
(France, cf Références), spécialisée dans la
sélection de cibles thérapeutiques à partir d’outils de la biologie intégrative
computationnelle, est de 70%!
Pour obtenir ce
résultat, Helios a construit un outil de saisie et de
construction de graphes d’interactions moléculaires. Ils ont aussi développé et
valider des algorithmes permettant de modéliser le fonctionnement des réseaux
d’interactions moléculaires avec notamment des réseaux complexes des 130
molécules.
L’utilité de la
biologie intégrative computationnelle dans l’identification de potentielles
cibles thérapeutiques a donc été prouvée.
Référence principale
Zhong, w. And Sterneberg P.W. (2007) Automated data integration for developmental biological research. Development 134, 3227-3238
Exemple d’application
de la biologie intégrative
Kristin C Gunsalus et al. (2005) Predictive models of molecular machines involved in Caenorhabditis elegans early embryogenesis. Nature 436, 861-865
Matériel
supplémentaire de l’article Gunsalus et al.
http://www.nature.com/nature/journal/v436/n7052/suppinfo/nature03876.html
Définition de biologie
intégrative
http://www.rhone-alpes-genopole.com/index.php?pageID=104&tabNum=3
Interactome utilisé par Gunsalus
Li, S. et al. (2004) A map
of the interactome network of the metazoan C.
elegans.
Science 303, 540-3
Matthews, L. R. et al.
(2001) Identification of potential interaction networks using
sequence-based
searches for conserved protein-protein interactions or "interologs".
Genome Res 11, 2120-6.
Présentation du projet
GO (Gene Ontology)
http://en.wikipedia.org/wiki/Gene_Ontology
http://artist.inist.fr/article.php3?id_article=261
GFP
http://fr.wikipedia.org/wiki/GFP
Technique
Double-Hybride
http://en.wikipedia.org/wiki/Two-hybrid
Helios-bioscience