GUIRAND
Jean-Daniel
JASSERAND
Maxime
Analyse
de recombinaison et de transferts horizontaux de gènes chez SARS-CoV-2


Avril 2021
Table des matières
Introduction et mise en
contexte
Historique génétique de SARS-CoV-2
· Présentation des nouvelles fonctionnalités
Introduction et mise en contexte
L’étude
porte sur l’analyse de la recombinaison et du transferts horizontaux de gènes
chez SARS-CoV-2 afin de déterminer son historique génétique.
Pour cela
nous allons utiliser des séquences ADN d'espéces
présentes dans l'arbre phylogénétiques de SARS-CoV-2 que nous allons analyser avec
le logiciel Simplot++. Nous
interpréterons ensuite les résultats pour tenter de répondre à cette
problématique.
L’arbre
phylogénétique ci-dessous nous permet de voir les différentes séquences qui
sont mises à l’étude :
Les quatre
premières séquences sont des séquences de SARS-CoV-2 humaines.
La séquence
du génome RatG13, en vert, présente une similarité avec celle du SARS-CoV-2.
En rouge la
séquence du génome Guangdong Pangolin et on a aussi
d’autres chauves-souris dont les gènes sont similaires à SARS-Cov-2.

Figure 1
- Arbre phylogénétique d'espèces de SARS-CoV-2
Historique génétique de SARS-CoV-2
· Hypothèses évolutives
Les
informations dont on dispose nous conduisent à trois hypothèses évolutives.
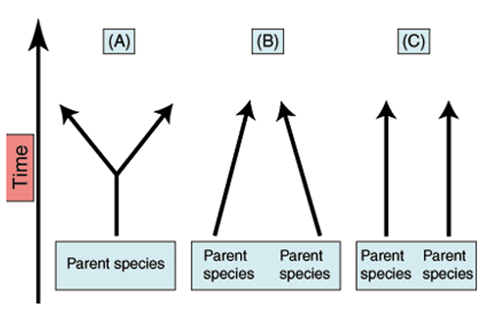
Figure 2
- Hypothèses évolutives
La première
connue sous le nom d’évolution parallèle (C) stipule que l'évolution parallèle
des 2 espèces aurait mené à des traits communs. Le problème avec cette
hypothèse est qu'elle ne peut pas expliquer pourquoi le RatG13 ne donne pas un
domaine RB similaire à SARS-CoV-2.
Cette
hypothèse est à rejeter, elle ne tient pas vraiment la route.
L’hypothèse
de l’évolution divergente (A) stipule que le GD pangolin, le SARS-CoV-2
auraient des ancêtres communs c’est ce qui expliquerait leurs similarités.
Selon cette hypothèse, il n’y aurait pas de recombinaison entre SARS-CoV-2 et le
pangolin de Guangdong. On ne rentre pas dans les détails dans le cadre de ce
rapport.
La recombinaison
génétique (B) est l'hypothèse sur laquelle nous allons élaborer. Elle
suggère qu’il y aurait recombinaison des gènes entre le pangolin de Guangdong
et SARS-CoV-2.
· Recombinaison génétique
Quand on
parle de recombinaison, deux modèles sont à considérer, à mentionner :
Le transfert
complet ou recombinaison intergénique. Le gène va être acquis dans son
entièreté par le génome receveur, l’arbre phylogénétique va être modifié si on
a vraiment un transfert complet. On parle de "recevoir" le gène en
entier du receveur au donneur. Cela peut être détecté facilement, dans le
premier gène on peut gérer toutes les séquences en établissant les similarités
entre chacune et trouver celles qui sont les plus proches et du coup établir un
transfert complet.
Le deuxième modèle dit transfert partiel est plus difficile
à analyser dans la mesure où une partie seulement du gène receveur-donneur va
être incorporé dans le génome du receveur ce qui fait qu’à l’intérieur d’un
même gène on peut avoir différentes sous-séquences qui ont des histoires
évolutives différentes.
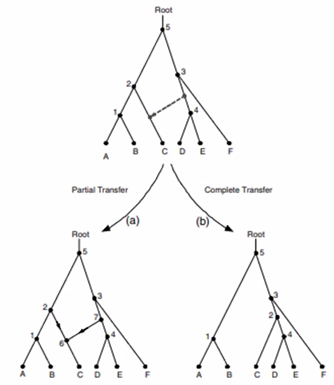
Figure 3
- Transferts partiel et complet
On parle alors de formation de gènes mosaïque et
recombinaison intra génique. Nous allons donc chercher à déterminer les
transferts partiels ou complets que SARS-CoV-2 a pu subir. Pour ce faire, on
utilisera le logiciel Simplot++ que nous allons
présenter dans la partie suivante.
A l'origine, Simplot est un logiciel créé en 1998 par Stuart Ray, dans le but d'effectuer des analyses de recombinaisons de gènes. Ce logiciel est encore aujourd'hui très utilisé par les chercheurs.
Malheureusement il n'est plus maintenu depuis 2003 et certains points pourraient être grandement améliorés.
Le principal est que l'application fonctionne sur Windows XP. Bien que les nouveaux OS soient capables de l'exécuter, l'application ne tire pas partie de leurs performances et le développement d'une interface plus moderne ne pourrait lui faire que du bien.
Figure 4 - Interface Simplot
Stéphane Samson, étudiant en maitrise, a donc décidé de faire de la refonte de ce logiciel son projet de recherche. Son objectif est de réimplémenter les fonctionnalités déjà présentes, en corrigeant les différents bugs, en améliorant l'interface, mais aussi d'ajouter de nouvelles fonctionnalités.
Tout d'abord, nous allons étudier le fonctionnement global de Simplot puis nous aborderons les nouvelles fonctionnalités apportées dans Simplot++. Nous conclurons en analysant nos séquences ADN pour répondre à la problématique.
·
Fonctionnement global
L'objectif de Simplot est de comparer des séquences d'ADN ou d'acides aminés pour trouver des similarités et ainsi détecter des recombinaisons ou des transferts horizontaux de gènes.
Pour cela, l'utilisateur va tout d'abord créer des groupes à partir de plusieurs séquences similaires afin que l'algorithme créer une séquence consensus pour chaque groupe.
Ensuite les séquences consensus à comparer vont être parcourues par fenêtre. On parle de technique de fenêtre coulissante (slinding window). Sur un même intervalle de positions, le programme va comparer un certain nombre de caractères entre chaque séquence consensus pour obtenir la similarité de cette fenêtre.
Les comparaisons sont effectuées en suivant des modèles de substitution. Leur rôle est de pouvoir calculer la similarité en suivant les lois biologiques de manière plus ou moins fidèle en appliquant différentes corrections.
Ainsi, avec le modèle de Hamming, le plus simple, le programme va uniquement compter les caractères similaires. D'autres modèles, comme Jukes-Cantor, vont prendre en compte le fait que les taux de substitution des nucléotides ne sont pas égaux. Pour finir, le modèle Kimura prend également en compte les taux de substitution mais va aussi s'intéresser aux nombres de transversion et translation.
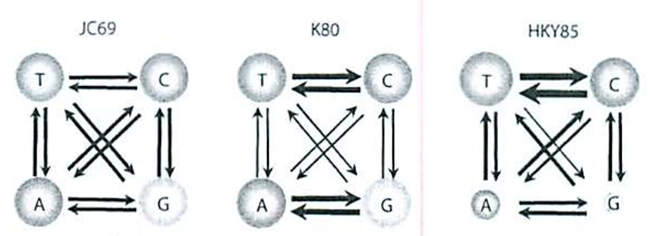
Figure 5 - Taux de
substitution entre différents modèle, une ligne plus épaisse indique des
chances plus élevées
La plupart du temps, un modèle plus réaliste aura un temps de calcul plus long, ce qui peut avoir une forte répercussion sur l'exécution du programme étant donné la longueur de nos séquences. Les différents modèles nous permettent d'avoir un éventail de possibilités et de trouver un compromis entre fiabilité et temps de calcul.
Après avoir calculé la similarité de notre fenêtre, le programme va la décaler d'un pas, un nombre de caractères paramétrable, puis réitérer la comparaison jusqu'à avoir parcouru la totalité de nos séquences.
Pour finir les pourcentages de similarité de chaque fenêtre seront affichés dans un graphique. Nous pourrons ainsi visualiser les séquences ayant la plus grande similarité globale ou encore constater des similarités locales sur certains gènes uniquement.
Figure 6 - Comparaison de
séquences
·
Présentation des nouvelles fonctionnalités
Un des objectifs dans la conception de Simplot++ est l'intégration d'une multitude de nouveaux modèles, afin que les utilisateurs puissent avoir un maximum de choix lors de leurs analyses. Nous passons ainsi de moins de 10 modèles à une vingtaine.
La fenêtre Network, inexistante sur le premier Simplot, permettra aux utilisateurs de visualiser les similarités sous forme d'un réseau au lieu d'un graphique.
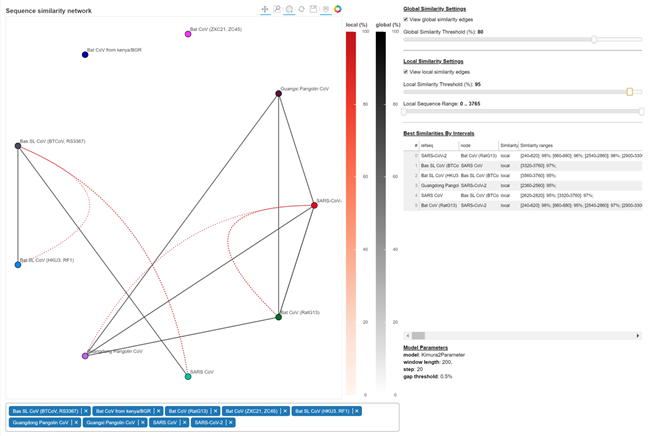
Figure 7 - Module Network
Ici par exemple, nous affichons une ligne droite noire entre les groupes dont la similarité globale dépasse 80% et une ligne courbe pointillée si la similarité local dépasse 95%.
Nous avons donc accès aux informations de manière plus complète et user-friendly, tout en pouvant manipuler les paramètres en temps réel.
Rapport de qualité et gestion d'erreur
Un défaut de la première version était l'absence de gestion d'erreur, si une séquence comportait trop de caractères manquants par exemple.
Dans Simplot++, la gestion d'exception permet en premier lieu d'empêcher le logiciel de crash et de prévenir l'utilisateur dans certain cas. En outre, la création d'un module de visualisation de la qualité permet de constater la fiabilité de l'analyse et de comprendre pourquoi une analyse peut défaillir.
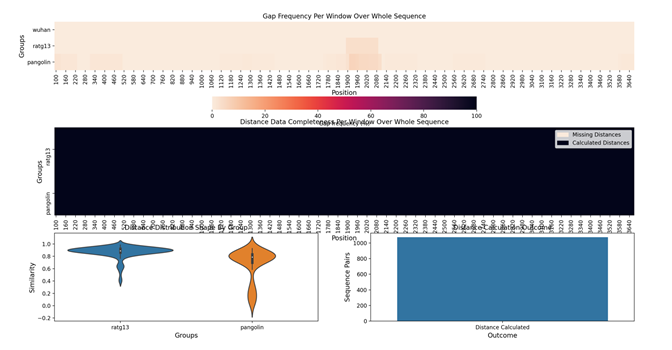
Figure 8 - Module qualité
Il est important de fournir aux utilisateurs un moyen de juger la qualité de leurs analyses.
Application
En utilisant Simplot++ nous avons pu obtenir les courbes de similarité suivantes :
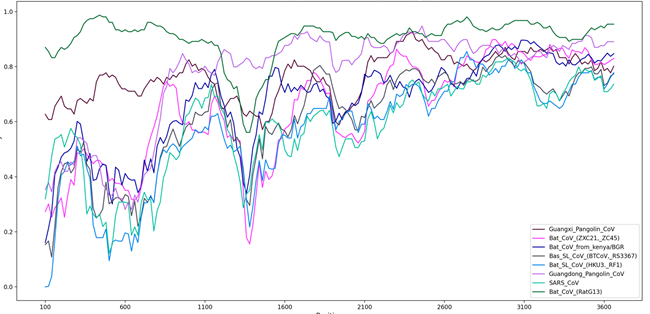
Figure 9
- Courbes de similarités de différents ADN par rapport à SARS-CoV-2
Nous pouvons constater que sur la grande majorité de la séquence, RatG13, l'ADN de chauve-souris, est la séquence la plus similaire, avec une similarité allant de 80% jusqu'à plus de 90%.
Toutefois, on remarque un changement notable entre les positions 1100 et 1600, correspondant au gène S, où le Pangolin de Guangdong devient similaire à 80% tandis que toutes les autres similarités baissent.
Nous pouvons donc déduire qu'un transfert horizontal de gènes a eu lieu sur cette portion.
Pour répondre à notre problématique, il semblerait que SARS-CoV-2 soit issu d'un transfert partiel entre du pangolin de Guangdong sur la chauve-souris RatG13
Références
https://sray.med.som.jhmi.edu/RaySoft/simplot_old/Version1/SimPlot_Doc_v13.html
https://www.cs.rice.edu/~nakhleh/COMP571/Slides/Phylogenetics-DistanceMethods-Full.pdf