Introduction
Nanopore is a technology of 3rd generation sequencing. It follows the two previous generations of sequencing technologies which are the traditional Sanger sequencing (1st generation) and the still very prevalent next generation sequencing (NGS) (2nd generation). Third generation sequencing is characterized by the possibility of sequencing long and ultra-long reads directly on nucleic acids, without requiring an amplification step. The new generation of instruments able to do this has become available fairly recently with the commercial release of the RS PacBio (Pacific Biosciences) in 2010 and the Nanopore MinION (Oxford Nanopore Technology) in 2014. In addition to sequencing long DNA molecules, Nanopore can also read RNA molecules directly, opening new avenues in our understanding of the transcriptome.
NGS is widely used nowadays for many applications and has been transformative in the study of genomics and transcriptomics, but it has major drawbacks. First, it can process only small size fragments and therefore requires a reference genome for alignment in order to identify the location of the sequencing reads. The possibility of getting long reads with 3rd generation sequencing facilitates greatly the work on organisms who do not have yet a reference genome, as it makes possible by the alignment of overlapping reads to perform de novo assembly. The short reads also complicate the sequencing of regions with repetitive sequences, as it hard to align reads of repetitive sequences to the genome. For RNA sequencing, they also limit the ability to identify which specific transcripts isoforms are expressed, because short reads will rarely cover several exon-exon boundaries to identify unambiguously what version of a transcript is being sequenced.
Moreover, NGS requires an amplification step of the starting material. This leads to some regions that hard to amplify (often GC-rich) to be underrepresented in the final coverage of the genome or the transcriptome. Another big issue is our understanding that genetic complexity of organisms resides in big part outside of the primary sequences of DNA or RNA molecules, but rather is associated with epigenetic and epitranscriptomic modifications of those molecules. The amplification step in NGS leads to the loss of such information prior to sequencing. Now, with a technology capable of analyzing directly DNA and RNA molecules as recovered from the cells, Nanopore constitutes a new tool that overcomes the drawbacks of NGS and could revolutionize our understanding of secondary modifications of nucleic acids and associated regulations.
Origins of the Nanopore technology
The principle behind the technology is that the passage of a single-stranded DNA (ssDNA) or RNA molecule through a nanoscale pore embedded into a membrane submitted to a voltage leads to small changes in the current that can then be decoded in order to identify the sequence of the DNA or RNA coming through. The idea is not new, as work on proteins potentially usable as biosensors has been done in multiple laboratories since the 1990s. Several obstacles had to be overcome in order to use it for actual sequencing, including the issues of resolution to distinguish bases from each other and speed of strand translocation through the pore (Figure 1).
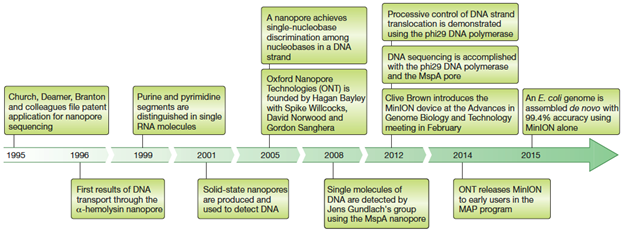
Figure 1: Main events in DNA nanopore sequencing history (Deamer et al. 2016).
As soon as 2005, it was demonstrated that all 4 types of bases could be distinguished from each other for a DNA molecule immobilized in an a-hemolysin pore, based on their effect on the ion conductance of the pore (Ashkenasy et al. 2005). The Oxford Nanopore Technology (ONT) company was founded around that same period (Figure 1). The transition to the MspA as biosensing pore in 2008 helped with the resolution by decreasing the size of the sensing region and therefore the number of nucleotides that are present and therefore contributing to the signal at any given time in a pore (Figure 2).
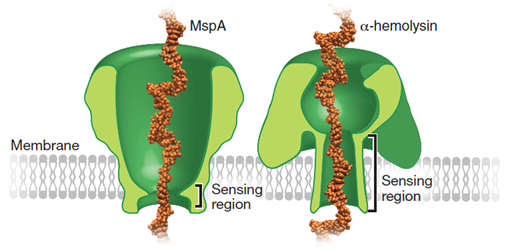
Figure 2: Proteins used as biosensor for the Nanopore technology. The sensing region in MspA is much shorter (4 nt) than for the a-hemolysine (12-15 nt), facilitating base calling for the nucleotides passing through it (Deamer et al, 2016).
The use of the phi29 DNA polymerase to optimize DNA unwinding and translocation speed through the pore was a key achievement towards getting a functional technology. As 4 nt are present in the sensor region at any given time, one of the first functional systems in 2014 was provided each possible 254 quadromers in order to identify the associated current signature and generate a “quadromer map”. The latter that was then used to interpret the ~100 passages through a pore that were necessary to reconstitute the sequence of the complete 4,500 bp of the phi X 174 genome (Laszlo et al. 2014). At this stage of the technology, many reads of the same molecule were still necessary because the 1-5ms time per base was not enough to get a clear signal given the size of the sensor and because the very small difference between bases on their effect on current inside the pore (H. Xing seminar). Yet, the first de novo assembly with a 99.4% accuracy of the Escherichia coli genome soon followed in 2015 (Loman et al. 2015) and many advances in algorithms to interpret the current signal for base calling were made since then with the help of machine learning.
The Oxford Nanopore Technology (ONT) currently available
Three main devices currently available differ mainly by their different ranges of sequencing capacity: MinION, GridION and PromethIon. They function all with the same basic flow cell (Figure 3) present in different number: 1, 5 or 24/48 flow cells, respectively.

Figure 3: Flow cell of the MinION. Nanopore sequencing in 2021 uses the same flow cell in all its devices. There are 512 channels in the sensor chip, each of them contains 4 pores.
With the present technology, the molecules (RNA or ssDNA) are brought to the pore bound to a motor protein that controls their speed through the nanopore (Figure 4). They move electrophoretically through the sensor at a rate of 30 bases per second (Deamer et al. 2016) and many algorithms exist and are still being developed to interpret the current signal into bases and specific bases modifications.
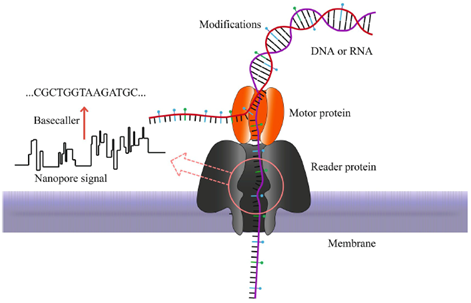
Figure 4: Pores function with a motor protein to control the denaturation of DNA and the speed of passage through a MSpA pore (H. Xing seminar).
Bioinformatics of Nanopore sequencing
Since the MinION was launched, numerous algorithms and tools have been developed to analyze the very long but highly error prone Nanopore data. These algorithmic solutions were generated to accomplish the main steps of data analysis such as base calling, data handling, read mapping, de novo assembly and variant discovery (see the main steps of nanopore data analysis workflow in the Figure 5.)
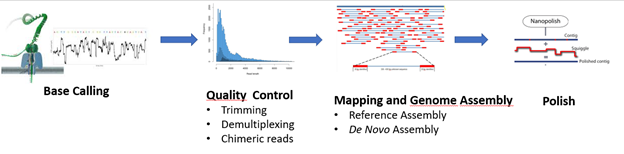
Figure 5: Nanopore sequencing analysis pipeline according to Oxford Nanopore Technologies.
In this report, we will briefly discuss the main steps of data analysis and will bring some separate examples of recent algorithms that have been shown to improve the accuracy of Nanopore sequencing.
Base calling
Base calling is the process of transforming a raw signal – for Nanopore, electric signals – obtained from a sequencer into a sequence of nucleotides. The accuracy of base calling is influenced by the chemistry used (the type of nanopore) as it can affect the signal-to-noise ratio. If this ratio is too low (the signal is too noisy), the determination of underlying sequence is virtually impossible (Rang et al. 2018). A second factor influencing the accuracy of base calling is the actual algorithm used for base calling. As mentioned above, base calling is a particular challenge in Nanopore sequencing because more than one base is read at any given moment in the pore (Figures 2 and 4). To discriminate between signal and noise and properly interpret the base composition, a software needs to be trained. However, a specific training dataset that is used may not be optimal if the DNA molecule being sequenced has, for instance, a strong nucleotide composition bias that the software has not been exposed to. As a result, training sets for new algorithms tend to diversify the type of sequences used as input. Zheng et al. (2020), for example, used reads performed by MinION for a variety of sequences such as the lambda phage and E. coli genomes, as well as human genomic DNA sequencing data released by the Nanopore WGS Consortium. For some users, it can be useful to develop custom trained models to improve base-calling accuracy and speed (Wick et al. 2019).
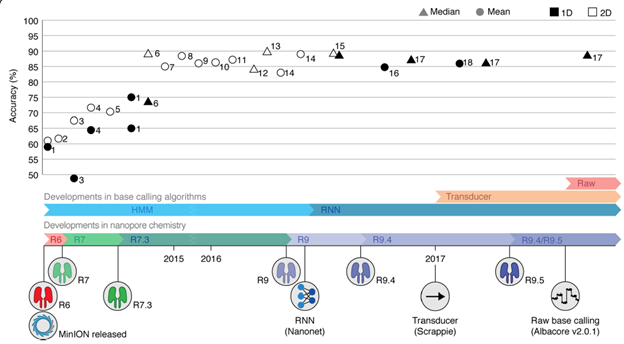
Figure 6: Timeline of chemistry changes and base-caller algorithms for Nanopore sequencing and the associated improvement in accuracy for base-calling (Rang et al. 2018). HHM Hidden Markov Model, RNN Recurrent Neural Network
Historically, there were 2 stages in the development of Nanopore base callers. The original ONT base caller used Hidden Markov Models (HMM) approach, but all current programs (since DeepNano) use Recurrent Neural Networks (RNN). Most recently, a Temporal Convolutional Network (TCN) approach (Causallcall) has been proposed that is said to be more accurate and faster that more traditional RNN approaches (Zheng et al. 2020). The base callers recommended by ONT (like Albacore and Guppy) are closed source, which limits the user’s ability to evaluate their base calling strategy. Some of the popular base callers developed for Nanopore sequencing are: Albacore, BasecRAWller, Chiron, DeepNano, Flappie, Guppy, Metrichor, Nanocall, Scrappie (reviewed in Makalowski and Shabardina, 2019).
MinION data formats and handling
MiniKNOW is the software that runs on the host computer to which the MinION is connected. It is used to set up parameters and perform runs, data acquisition and real-time signal segmentation and feedback of experimental progression. For each read, the signal segmentation (segment mean, variance and duration) and the metadata associated are stored by the MinKNOW in FAST5 binary files, a variant of the HDF5 standard. The data generated is then converted from FAST5 files into the more traditional FASTA or FASTQ sequence formats that can then be used by other softwares (Magi et al. 2017).
Sequence assembly
The purpose of de novo genome assembly is to reconstruct the entire genome sequence from a collection of sequenced reads much shorter than the genome from which they are generated. Indeed, even long reads (the longest recorded DNA read is 2.3 Mb) such as those produced by Nanopore still require assembly to reconstitute a complete genome. From the very beginning of sequencing, an assembly of raw reads has been used in order to obtain complete, contiguous sequences. Originally, sequence assemblers used overlaps to merge and order raw sequences, an approach called Overlap-Layout-Consensus (OLC). Although not appropriate to align short reads of NGS, this approach is once again being used for the alignment of long reads resulting from 3rd generation sequencing. One such an assembler is Canu, based on Celera Assembler, which has been used for a successful assembly of a variety of genomes of bacteria, fungi, insects, plants and fish. Many assemblers exist for Nanopore’s long but low accuracy reads: ABruijn, Canu, Flye, Miniasm, NanoPipe, Racon, wtdbg2, etc. (reviewed in Makałowski and Shabardina, 2019).
Variant detection
Reference genomes are usually constructed from a consensus sequence based on several individuals. But of course, each individual member of a given species is different at the genome level. Variations can be also acquired somatically during malignant transformation. Identifying those variations in the genome can be one of the primary reasons for genome sequencing. Considering its rather low accuracy (at least compared to NGS technologies), Nanopore is not necessarily the best suited tool to identify genetic variants consisting of SNPs, or small insertions or deletions (Indels), although Nanopore sequencing can achieve very good accuracy for the very deep sequencing of shorter genomes. It can however be very performant in identifying structural variants, such as large Indels or complex rearrangements, including translocations and inversions, which tend to be invisible by NGS.
Still, statistical methods have been developed to evaluate the likelihood that observed differences between Nanopore reads and a reference sequence may be variants rather than sequencing artifacts. Different approaches exist for variant calling. While most programs try to identify variants after base calling and often by comparing a consensus sequence with a reference, some, like Nanopolish, are using raw signal information. Here are some commonly used softwares for variants identification: Clair, HapCut2, Medaka, NanoPipe, Nanopolish, and Sniffles (reviewed in Makałowski and Shabardina 2019).
New suggested workflow for direct RNA sequencing
While the traditional workflow is widely accepted, improvements are constantly proposed. Recently, a new pipeline named MasterOfPores was suggested by Cozzutto et al. 2020 for the analysis of direct RNA sequencing reads (Figure 7). Raw current intensities are first converted into multiple types of processed data such as FASTQ and BAM, providing information on the quality of the run, base calling and mapping of the reads. Further analysis is then performed on mapped reads such as computation of per-gene counts, prediction of RNA modifications, identification of polyA tail length and RNA isoforms. The MasterOfPores is freely available in GitHub (https://github.com/biocorecrg/master_of_pores). One of the purposes of this workflow is to significantly simplify the analysis of nanopore direct RNA sequencing data by non-bioinformatics experts, thus improving the understanding of the epitranscriptome.
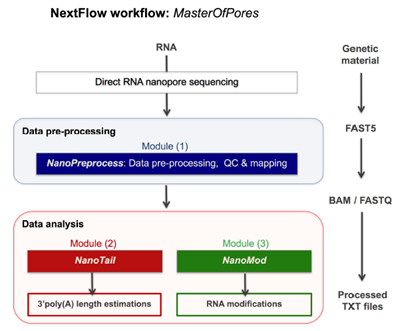
Figure 7: Overview of the MasterOfPores workflow for the processing of direct RNA nanopore sequencing datasets. The workflow accepts both single FAST5 and multi-FAST5 reads and includes 5 main steps: i) base-calling, ii) filtering, iii) quality control, iv) mapping and v) final report building (Cozzutto et al. 2020).
Current applications of Nanopore technology
Many publications are generated using this technology. The Dimensions website counted 28,479 publications featuring the keyword “nanopore sequencing” as of Mar 4th 2021, including more than 1100 already in 2021. Without even trying to be exhaustive, here are some of the recent applications that we could identify and that illustrate the variety of potential applications.
As mentioned earlier, the possibility to investigate directly nucleic acids without amplification is a great opportunity to investigate the epigenome and other DNA modifications. The most common type of epigenetic change is the methylation of cytosine (5-methylcytosine), which is involved in many biological processes, such as gene regulation and cell differentiation and a lot of work has been done to be able to study this type of modification. Two different software, Nanopolish and SignalAlign, are using HMMs to identify 5-methylcytosine with high accuracy. A more recent addition, DeepMod, uses raw electric signals and a bidirectional RNN to detect 5-methylcytosine and N6-methyldeoxyadenosine, which are both forms of epigenetic modifications (Liu et al. 2019).
Another promising application of the Nanopore sequencing lies within metagenomic studies where one attempts to identify the multiple species of microorganisms present in a complex sample. Species are classically identified through their rRNA sequence, but rRNA tend to be a very conserved molecule and short reads often cannot distinguish between closely related species or microbial strains. Long reads, however, are a potential solution to that problem by allowing the analysis of the whole rRNA sequence, including the less conserved regions that allow species identification. A few dedicated softwares have been developed for that kind of use, including MetaG and Nanopore’s EMPI2ME (Makałowski and Shabardina 2019).
In view of the recent pandemic of COVID-19, ONT has been actively developing a novel tool called LamPORE for the diagnostic of SARS-CoV-2 infection. It consists of a complete workflow adapted to the GridION system based on the sequencing of 2 conserved viral genes. It starts from a purified RNA sample and ends with a completely automated analysis that requires no human intervention or interpretation.
There is an infinity of potentially useful way to use the Nanopore technology for human health. Thanks to its great transportability, ease of use, and real-time availability of the sequencing results, Nanopore’s MinION is becoming a great tool to identify unknow organisms in a time sensitive manner, including microorganisms relevant to human health. Although the standard method requires 2 to 5 days, Nanopore sequencing has recently been used to successfully identify pathogens and antimicrobial resistance genes extracted from positive blood culture, all that after only 10 min of sequencing (Taxt et al. 2020).
Conclusion and perspectives
Nanopore sequencing has been long in the making, but with some great advances in pore technology, as well as in the bioinformatics methods necessary to interpret and use the data, it is now a very useful tool with an infinity of potential applications, be it the genome sequencing of new species or the investigation of the newly discovered epitranscriptome.
The material used in sequencing chips is still of biological source with pores from bacterial origin bound to a bilipidic membrane. They have the advantage of being cheap to produce and easy to modify as needed, but they create constraints regarding the condition of their use in term of usable voltage, or available diameter. They also create a signal that is fairly noisy. But there are currently efforts to replace them with graphene nanopores (constituted of a single layer of atoms) on a silicone membrane that would not suffer from those limitations and could be mass produced. They would also improve the capacity for accurate base calling by allowing possibly the measurement of a single base at a time.
Beyond potential technical improvements of the chips, some of the most promising application of the technology Nanopore have to do with the investigation of epigenetic and epitranscriptomic modifications of nucleic acids. Advances in this field will come mostly from the development of new bioinformatics tool to interpret chemical changes to bases that cause very subtle effects in current pattern, making them even harder to identify from the noise of the technique. The resulting knowledge promises to revolutionize our understanding of genes and gene expression.
References
Ashkenasy N, Sánchez-Quesada J, Bayley H, Ghadiri MR. Recognizing a single base in an individual DNA strand: a step toward DNA sequencing in nanopores. Angew Chem Int Ed Engl. 2005 Feb 18;44(9):1401-4. doi: 10.1002/anie.200462114.
Cozzuto L, Liu H, Pryszcz LP, Pulido TH, Delgado-Tejedor A, Ponomarenko J, Novoa EM. MasterOfPores: A Workflow for the Analysis of Oxford Nanopore Direct RNA Sequencing Datasets. Front Genet. 2020 Mar 17;11:211. doi: 10.3389/fgene.2020.00211.
Deamer D, Akeson M, Branton D. Three decades of nanopore sequencing. Nat Biotechnol. 2016 May 6;34(5):518-24. doi: 10.1038/nbt.3423.
Laszlo AH, Derrington IM, Ross BC, Brinkerhoff H, Adey A, Nova IC, Craig JM, Langford KW, Samson JM, Daza R, Doering K, Shendure J, Gundlach JH. Decoding long nanopore sequencing reads of natural DNA. Nat Biotechnol. 2014 Aug;32(8):829-33. doi: 10.1038/nbt.2950.
Liu Q, Fang L, Yu G, Wang D, Xiao CL, Wang K. Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat Commun. 2019 Jun 4;10(1):2449. doi: 10.1038/s41467-019-10168-2.
Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods. 2015 Aug;12(8):733-5. doi: 10.1038/nmeth.3444.
Magi A, Semeraro R, Mingrino A, Giusti B, D'Aurizio R. Nanopore sequencing data analysis: state of the art, applications and challenges. Brief Bioinform. 2018 Nov 27;19(6):1256-1272. doi: 10.1093/bib/bbx062.
Makałowski W, Shabardina V. Bioinformatics of nanopore sequencing. J Hum Genet. 2020 Jan;65(1):61-67. doi: 10.1038/s10038-019-0659-4.
Rang FJ, Kloosterman WP, de Ridder J. From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018 Jul 13;19(1):90. doi: 10.1186/s13059-018-1462-9.
Taxt AM, Avershina E, Frye SA, Naseer U, Ahmad R. Rapid identification of pathogens, antibiotic resistance genes and plasmids in blood cultures by nanopore sequencing. Sci Rep. 2020 May 6;10(1):7622. doi: 10.1038/s41598-020-64616-x.
Wick RR, Judd LM, Holt KE. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019 Jun 24;20(1):129. doi: 10.1186/s13059-019-1727-y.
Zeng J, Cai H, Peng H, Wang H, Zhang Y, Akutsu T. Causalcall: Nanopore Basecalling Using a Temporal Convolutional Network. Front Genet. 2020 Jan 20;10:1332. doi: 10.3389/fgene.2019.01332.