Nicolas de Montigny
Abdellatif El Ghizi
Séminaire interdisciplinaire
de bio-informatique – BIF 7002
Rapport de conférence –
Zahia Aouabed
Travail présenté à
Pr. Abdoulaye Banire Diallo
et
Pr. Vladimir Makarenkov
Département
des sciences
Université du Québec à Montréal
Remis le : 18 février 2020
Introduction
Contrairement à la
génomique qui est basée sur l’isolement et la culture d’un génome donné, la
métagénomique est une discipline émergente qui consiste à étudier l’ensemble
des génomes présents dans un milieu par séquençage à haut débit et analyse de
l’ensemble de l’ADN de ce milieu. Le terme métagénomique a été proposé pour la
première fois par Handelsman et al en 1998 (1) mais son application n’a émergé que lors des dernières années avec la
généralisation des techniques de séquençage haut débit.
En effet, les récents progrès en termes de technologies
de séquençage haut débit, le développement de nouvelles méthodes
bio-informatiques ainsi que le BIG-data ont permis des avancées importantes
dans la métagénomique. Ainsi, son champ d’application a été élargi (santé,
agro-alimentaire, écologie, foresterie, etc) et des projets de recherches
d’envergure ont été menés à échelle internationale (2).
Toutefois,
des défis techniques et conceptuels sont à relever. Ces défis sont liés
principalement à l’analyse, l’exploitation et la qualité des données
métagénomiques, car le séquençage d’un génome n’est pas applicable à tous les
organismes. De plus, les séquenceurs « Next Generation Sequencing » (NGS)
ont besoin d’une quantité importante de matériel génétique pour pouvoir
produire des ensembles des données qui couvrent le génome dans son ensemble (3).
La
métagénomique s’intéresse principalement à l’étude des microbiomes, mais peut
aussi s’appliquer à l’étude de populations où il y a présence d’eucaryotes (ex :
zooplancton dans un lac). Pour les eucaryotes il n’y a pas de problème au
séquençage, car ces organismes multicellulaires peuvent produire de grandes
quantités d’ADN. Cependant, pour les procaryotes cela peut poser problème
puisque ce sont des organismes unicellulaires possédant de petits génomes. Il
faut donc soit faire des cultures pour dupliquer ces cellules, sachant que la
majorité de ces organismes ne peuvent survivre en milieux contrôlé, ou utiliser
la technique « Single Cell Sequencing » (sc-seq). Cependant, l’inconvénient de
cette dernière est qu’il peut y avoir certaines régions du génome qui sont
favorisés : seulement 40% du génome est couvert en moyenne à la suite de l’amplification
par PCR des gènes (3).
Types d’analyse des données
métagénomiques
L’analyse de données métagénomiques cible
donc une communauté et cherche à caractériser cette communauté afin de pouvoir
faire des analyses différentielles entre plusieurs conditions, par exemple, ou seulement
d’en apprendre plus sur une communauté. Il existe ainsi quatre types d’analyse
des données métagénomiques possibles (3, 4) :
1-
L’analyse taxonomique :
un inventaire des taxons ou espèces présentes dans le milieu et leurs abondances.
2-
Assemblage
des différents génomes : assemblage partiel ou global des génomes d’espèces
présentes dans le milieu selon la technique de séquençage utilisée.
3-
L’analyse fonctionnelle :
permet de déterminer la fonction d’une communauté dans son ensemble ou de
diviser cette communauté en sous-groupes fonctionnels interagissant ensembles.
Peut mener à la cartographie des voies métaboliques présentes dans une
communauté.
L’analyse
comparative : comparaison des données de plusieurs communautés provenant
de différents milieux du même type (ex : microbiome racinaire d’une espèce
d’arbre présente dans différents sols) ou conditions d’un même milieu (ex :
microbiome buccal avec et sans maladie).
Stratégies en métagénomique
Il est important
de pouvoir compter sur des techniques de séquençage ne se basant pas sur l’amplification
des séquences présentes et ne requérant pas de culture puisque la majorité des
microorganismes ne peuvent croitre en milieu artificiel. C’est pourquoi les
technologies tels le « Whole Genome Shotgun » (WGS) et le séquençage
ciblé sont si intéressants (2).
La métagénomique ciblée
Cette approche
consiste à l’amplification puis au séquençage d’une région ou portion
particulière du génome relativement petite en quelque centaines de paires de bases.
Cette région est appelée marqueurs conservés, généralement on utilise les RNAs ribosomaux
comme marqueurs. Pour les bactéries, il s’agit de l’ADN ribosomal 16S, qui est
un excellent marqueur phylogénétique et un taux de mutation relativement stable
puisque sa fonction est essentielle à l’organisme (2, 3).
Plusieurs
autres marqueurs sont possibles dépendamment de l’ordre taxonomique observé.
Ainsi pour les bactéries, on peut aussi utiliser ITS, CPN60 et RescA par
exemple. Pour les eucaryotes, on utilise principalement l’ARN 18S qui est l’équivalent
de l’ARN 16 des procaryotes et ITS. Pour les virus, on peut entre-autres
utiliser Gp23(bacteriophagesT4-like), RdRp (picornaviruses) (3).
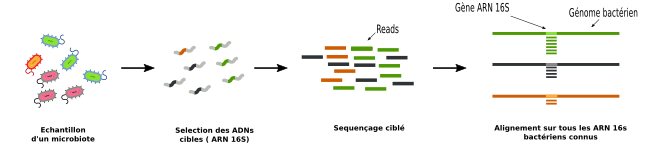
Figure 1 Stratégie ciblé : Seuls les ADNs du gène cible sont séquencés. Exemple
en bactériologie, le gène cible est l'ARN 16S (5)
À la
suite d’un séquençage, les « reads » appelés « Operational
Taxonomic Units » (OTU) sont regroupés par similarité. Ainsi, on peut
produire un graphique des groupes (« clusters ») afin de les
identifier à un taxon ou une fonction. Ces groupes peuvent être comparés ou non
à une référence selon trois stratégies : de novo où on ne compare
pas à une référence; closed-reference où on se base uniquement sur la
comparaison à des séquences connues; open-reference où on alie les deux
autres stratégies. Plusieurs techniques de « clustering » sont
possibles pour avoir des groupes plus ou moins rapprochés du centroïde (3, 6).
Selon Guyomar
et al, (7) , les deux approches de métagénomique WGS et ciblée
peuvent être utilisées conjointement. Elles sont complémentaires, notamment
dans le cas des communautés les plus complexes, où les organismes rares ne
peuvent être identifiés que par des méthodes ciblées
La métagénomique globale WGS
Comme
son nom l’indique, cette stratégie permet d’étudier la composition globale du microbiome
de l’échantillon environnemental. De plus, cette technique est effectuée sans
faire d’amplification préalable des génomes. L'ensemble des ADNs
du microbiote sont séquencés générant
ainsi une importante quantité de fragments géniques de tous les organismes
présents.
Un assemblage des séquences obtenues en les comparant par alignement sur des
génomes référencés permettent d’identifier les espèces connues formant le
microbiome. Les génomes inconnus assemblés de novo puis annotés et
incorporés dans des bases de données (3, 4).
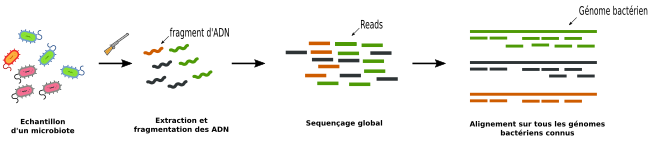
Figure 2 Stratégie globale : L'ensemble des ADNs présents dans un
échantillon de microbiote sont séquencés (5).
Il existe deux approches à la stratégie
WGS, la première est basée sur les « reads » et cherche à classifier
les « reads » dans un optique de taxonomie et de fonction. L’alignement
est donc fait avec des génomes de références dès le début et peut être pratique
pour la détection d’organismes d’intérêt. La deuxième stratégie est basée sur l’assemblage,
on y assemble tous les fragments de novo en groupements génomiques puis
ils sont comparés à des génomes connus (« binning »). Cette deuxième
stratégie permet la découverte de nouveaux génomes plus facilement (4, 6).
Outils principaux en métagénomique
La
métagénomique est une approche de plus en plus populaire dans plusieurs
domaines dont l’écologie, la biologie médicale, la criminalistique, etc. Il
existe donc de nombreux outils pour analyser et visualiser les résultats de
séquençage. De plus, puisque c’est une avenue de plus en plus populaire grâce à
l’accessibilité des NGS qui augmente, il y a souvent de nouveaux outils qui
sont développés. Ainsi on essaiera de présenter une liste exhaustive des outils
principaux pour les deux approches de la métagénomique. Certains outils sont
disponibles sur le web et possibilité d’installation locale tandis que d’autres
sont uniquement disponibles pour installation locale ou sur des serveurs de
calcul.
Outils spécialisés en métagénomique ciblée
Qiime2 :
Outils en ligne de commande permettant l’analyse de données brutes de
séquençage. Il permet aussi l’identification des OTU et la reconstruction
phylogénétique. De plus, il permet de produire des graphiques facilement et de
bonne qualité. (3, 8-10)
UPARSE :
Outils en ligne de commande permettant l’identification de clusters à partir de
données de séquençage d’ARN 16S.(8, 11)
Mothur :
Outils en ligne de commande visant à exprimer les variations environnementales.
Permet le clustering via l’identification d’OTU. (3, 8, 12)
DADA2 :
Package R permettant l’identification des OTU, la filtration, l’identification
de chimères et autres à partir de résultats d’amplicons ciblés.(8, 13)
MED :
Package R permettant l’utilisation de gènes cibles. Permet aussi d’intégrer les
résultats métagénomiques dans un optique écologique et de phylogénétique. (8, 14)
METAVIR2 :
Outils web permettant l’analyse de séquençage métagénomique uniquement pour les
virus. Fourni des arbres phylogénétiques pré-faits à partir de gènes cibles. (15, 16)
Outils spécialisés en métagénomique globale WGS
MataPhlAn2 :
Outils en ligne de commande permettant l’analyse du profil bactérien. Produit
des graphiques de grande qualité. (3, 8, 9, 17)
Kraken :
Outils en ligne de commande permettant l’identification taxonomique de
séquences d’ADN en utilisant l’alignement de k-mers. Il est disponible
en deux versions Kraken et mini-Kraken. Le second est plus rapide, mais prend
moins de paramètres en compte. (8, 18)
CLARK :
Outils en ligne de commande permettant la classification de séquences
nucléiques en se basant sur une classification supervisée et en utilisant un
jeu réduit de k-mers. (8, 19)
MOCAT :
Outils en ligne de commande proposant un « pipeline » rapide et
personnalisable. Permet la prédiction de gènes codant pour des protéines.
Compare les séquences à des bases de données et retourne les résultats sous
forme de fichier Excel et base de données SQL. (9, 20)
VIROME :
Outils web permettant l’analyse de données métagénomiques virales. La
classification est faite à l’aide des ORF en se basant sur les données connues
par le serveur de données et les séquences environnementales. Fourni une
interface graphique pour l’exploration et l’interprétation facilitée des
résultats. (16, 21)
Outils pouvant effectuer les deux types d’analyses
MG-RAST :
Outils web se basant sur d’autres serveurs et outils permettant le profilage fonctionnel
et la composition de communautés microbiennes. Permet de générer des graphiques
élaborés selon le besoin. Prend en charge les données métagénomiques brutes et
les analyse avec son propre « workflow » automatiquement en plus de
faire l’annotation des séquences. (8, 16, 22)
METAGENassist :
Outils web gratuit permettant l’analyse statistique multivariée des données
générées par d’autres outils d’analyse de métagénome. Peut produire un
consensus entre des microbiomes multiples ainsi qu’une cartographie phénotypique
des microbiomes. (16, 23)
EBI
Metagenomics : Interface web développé par l’EBI. Il utilise des outils de
Qiime et autres développés par l’EBI pour l’analyse des métagénomes. La base de
données utilisé est celle de l’EMBL-EBI pour comparaison de toutes les séquences
et permet l’intégration dans la base de données directement. L’outils permet aussi
de faire de contrôle de qualité et le « trimming » des séquences. (16, 24)
Limites de la métagénomique
Selon Siegwald (25), la métagénomique a plusieurs limites :
·
Approche descriptive :
la métagénomique permet de décrire et identifier les espèces présentes dans le microbiote
étudié. La compréhension du fonctionnement de la variation des microbiotes
reste un enjeu à appréhender et l’intégration des métadonnées relative à
l’écosystème devrait permettre d’orienter la métagénomique vers une direction
explicative et prédictive. (25, 26)
·
Absence d’une méthode
standard de référence et analyse des données générées : Tel que vu dans
les outils, il existe de nombreux « workflow » pour l’analyse des données
métagénomiques ce qui les rend difficile à comparer entre elles. De plus, il
n’existe aucun consensus sur l'approche analytique à mener dans un contexte métagénomique
malgré l’abondance de l’offre des logiciels d’analyses. Cela est dut au fait qu’il
n’existe pas une méthode ou un processus de référence à suivre pour mener un
projet métagénomique. (25, 27)
·
La sélection d'un locus
cible suffisamment discriminant entre les organismes d'intérêt, fortement
limitée par la courte taille des lectures de séquençage (< 500 nt). Au vu de
cette quantité d’information restreinte, la métagénomique ne permet ainsi pas
d'obtenir une image détaillée du microbiote d'intérêt, l’information contenue
dans la littérature n’étant pas souvent suffisante pour discriminer les espèces
entre elles. Le profilage phylogénétique pourrait être amélioré par
l'utilisation des 40 gènes essentiels au lieu de se focaliser seulement sur le
16S puisque cela entraine une diminution de la résolution des résultats. (25, 26)
Références
1. Handelsman J, Rondon MR, Brady SF,
Clardy J, Goodman RM. Molecular biological access to the chemistry of unknown
soil microbes: a new frontier for natural products. Chem Biol.
1998;5(10):R245-R9.
2. Sudarikov K, Tyakht A,
Alexeev D. Methods for The Metagenomic Data Visualization and Analysis. Current
Issues in Molecular Biology. 2017:37-58.
3. Aouabed Z.
Métagénomique et la bioinformatique des séquences. UQAM; 2020.
4. Jünemann S,
Kleinbölting N, Jaenicke S, Henke C, Hassa J, Nelkner J, et al. Bioinformatics
for NGS-based metagenomics and the application to biogas research. Journal of
Biotechnology. 2017;261:10-23.
5. Schutz S. Introduction
à la métagénomique Date inconnue [Available from: http://dridk.me/metagenomique.html.
6. Ghurye JS,
Cepeda-Espinoza V, Pop M. Metagenomic Assembly: Overview, Challenges and
Applications. Yale J Biol Med. 2016;89(3):353-62.
7. Guyomar C, Delage W,
Legeai F, Mougel C, C., Simon J-C, Lemaitre C. Reference guided genome assembly
in metagenomic samples. RECOMB 2018 -
22nd International Conference on Research in Computational Molecular Biology;
2018-04-21; Paris, France2018. p. 1.
8. Niu S-Y, Yang J,
McDermaid A, Zhao J, Kang Y, Ma Q. Bioinformatics tools for quantitative and
functional metagenome and metatranscriptome data analysis in microbes.
Briefings in Bioinformatics. 2017;19(6):1415-29.
9. Escobar-Zepeda A,
Godoy-Lozano EE, Raggi L, Segovia L, Merino E, Gutiérrez-Rios RM, et al.
Analysis of sequencing strategies and tools for taxonomic annotation: Defining
standards for progressive metagenomics. Sci Rep. 2018;8(1):12034-.
10. Kuczynski J, Stombaugh
J, Walters WA, González A, Caporaso JG, Knight R. Using QIIME to analyze 16S
rRNA gene sequences from microbial communities. Curr Protoc Bioinformatics.
2011;Chapter 10:Unit10.7-.7.
11. Edgar RC. UPARSE: highly
accurate OTU sequences from microbial amplicon reads. Nat Methods.
2013;10(10):996-8.
12. Schloss PD, Westcott SL,
Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur:
open-source, platform-independent, community-supported software for describing
and comparing microbial communities. Appl Environ Microbiol.
2009;75(23):7537-41.
13. Callahan BJ, McMurdie
PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample
inference from Illumina amplicon data. Nat Methods. 2016;13(7):581-3.
14. Eren AM, Morrison HG,
Lescault PJ, Reveillaud J, Vineis JH, Sogin ML. Minimum entropy decomposition:
Unsupervised oligotyping for sensitive partitioning of high-throughput marker
gene sequences. The ISME Journal. 2015;9(4):968-79.
15. Roux S, Tournayre J,
Mahul A, Debroas D, Enault F. Metavir 2: new tools for viral metagenome
comparison and assembled virome analysis. BMC Bioinformatics. 2014;15(1):76.
16. Dudhagara P, Bhavsar S,
Bhagat C, Ghelani A, Bhatt S, Patel R. Web Resources for Metagenomics Studies.
Genomics Proteomics Bioinformatics. 2015;13(5):296-303.
17. Truong DT, Franzosa EA,
Tickle TL, Scholz M, Weingart G, Pasolli E, et al. MetaPhlAn2 for enhanced
metagenomic taxonomic profiling. Nat Methods. 2015;12(10):902-3.
18. Wood DE, Salzberg SL.
Kraken: ultrafast metagenomic sequence classification using exact alignments.
Genome Biol. 2014;15(3):R46-R.
19. Ounit R, Wanamaker S,
Close TJ, Lonardi S. CLARK: fast and accurate classification of metagenomic and
genomic sequences using discriminative k-mers. BMC Genomics. 2015;16(1):236-.
20. Kultima JR, Sunagawa S,
Li J, Chen W, Chen H, Mende DR, et al. MOCAT: A Metagenomics Assembly and Gene
Prediction Toolkit. PLOS ONE. 2012;7(10):e47656.
21. Wommack KE, Bhavsar J,
Polson SW, Chen J, Dumas M, Srinivasiah S, et al. VIROME: a standard operating
procedure for analysis of viral metagenome sequences. Standards in Genomic
Sciences. 2012;6(3):421-33.
22. Meyer F, Paarmann D,
D'Souza M, Olson R, Glass EM, Kubal M, et al. The metagenomics RAST server - a
public resource for the automatic phylogenetic and functional analysis of
metagenomes. BMC bioinformatics. 2008;9:386-.
23. Arndt D, Xia J, Liu Y,
Zhou Y, Guo AC, Cruz JA, et al. METAGENassist: a comprehensive web server for
comparative metagenomics. Nucleic Acids Research. 2012;40(W1):W88-W95.
24. Hunter S, Corbett M,
Denise H, Fraser M, Gonzalez-Beltran A, Hunter C, et al. EBI metagenomics—a new
resource for the analysis and archiving of metagenomic data. Nucleic Acids
Research. 2013;42(D1):D600-D6.
25. Siegwald La. Solutions
to improve targeted metagenomics studies: Université du Droit et de la Santé -
Lille II; 2017.
26. Rogue M. La
métagénomique et les défis pour la bioinformatique au goût #IHMC2012 2012
[updated 2012/04/04. Available from: https://bioinfo-fr.net/la-metagenomique-et-les-defis-pour-la-bioinformatique-au-gout-ihmc2012.
27. Champomier-Verges
M-C, Zagorec M. La métagénomique : développements et futures applications:
Editions Quae; 2015 2015. 116 p.