UNIVERSITÉ DU QUÉBEC À MONTRÉAL
RAPPORT
Analyse de recombinaison et de transferts horizontaux de gènes chez SARS-CoV-2
Conférence de Stéphane Samson
PRÉSENTÉ À
VLADIMIR MAKARENKOV
COURS
BIF7002 – 30
Baloul Dihya, Marinelli Marina, Audrey-Ann Sicard
8 mars 2022
Trimestre d’hiver 2022
TABLE DES MATIÈRES
Introduction
Mise en contexte
La recombinaison génétique est un échange d'information génétique entre deux génomes, c’est un processus qui est répandu chez les virus et qui contribue à leur diversité. Ces recombinaisons permettent aux virus d’acquérir des fonctions leur permettant de surmonter la pression sélective et de s'adapter à de nouveaux hôtes et environnements [4]. La détection de ces transferts de gène qui se produisent au niveau d’un génome nous permettent de comprendre l’histoire évolutive des espèces et déduire ainsi leurs origines.
Depuis 2019, le monde fait face à un virus, le Covid-19, qui est considéré comme étant l’un des plus dangereux au monde. Depuis les premiers signalements de ce nouveau coronavirus ressemblant au Syndrome Respiratoire Aigu Sévère de 2003 (SARS-CoV-1) à Wuhan, en Chine, les chercheurs essaient de comprendre comment ce syndrome a pu franchir les barrières de l’espèce et devenir pathogène chez l’être humain. Depuis, le débat s'est concentré sur deux idées concurrentes: un scénario de fuite du laboratoire qui n’a pas pu encore être prouvé et une émergence zoonotique qui reste la seule explication possible à nos jours [4].
Selon les scientifiques, l’introduction d'un coronavirus épidémique hautement pathogène dans la population humaine est dû à d’importants transferts d’informations génétiques entre les différentes espèces de CoVs déjà existantes chez les espèces animales. Pour déterminer donc, l’histoire évolutive de ce virus et identifier l’origine de la maladie, plusieurs études basées sur des analyses phylogéniques des génomes du Covid-19 et ceux d’autres coronavirus ont été effectuées donnant ainsi plusieurs hypothèses sur son évolution [9], [8], [10], [12]. Bien que les scientifiques n’arrivent pas à aboutir à une conclusion sur le potentiel hôte intermédiaire du SARS-CoV-2, n’est au moins, ils sont tous d’accord sur le fait que la chauve-souris et son hôte naturel. Ils sont aussi d’accord sur la possibilité de l’émergence de ce virus à la suite de recombinaisons et transferts de gènes qui ont pu se produire entres différentes souches de CoVs déjà existantes chez des espèces d’animaux.
L’origine du SARS-COV-2
La recherche sur l’origine du Sars-CoV-2 a abouti à l’idée que, tout comme SARS-CoV-1, c’était un coronavirus hébergé par une ou plusieurs espèces de chauve-souris qui étaient à l’origine du coronavirus humain (chauve-souris réservoirs de virus) [13]. SARS-CoV-2 appartient au β-coronavirus, avec un génome hautement identique à celui du coronavirus de la chauve-souris. Ce qui indique que la chauve-souris est peut-être son hôte naturel [3].
Bien que le SARS-CoV-2 présente une similarité de 96% avec celui de la chauve-souris, la différence observée au niveau de la région codant pour la protéine S laisse penser que ce virus n’a pas pu se transmettre directement de la chauve-souris à l’homme, mais plutôt, à travers au moins une espèce intermédiaire dont on ignore exactement l’identité. La consommation d'animaux sauvages ou le contact direct avec ces hôtes intermédiaires ont été suspectés d'être un mode de transmission initial [10].
Des études ont montré que le SARS-CoV-2 présente aussi une grande ressemblance dans certaines régions avec différents génomes de coronavirus de pangolins malais [10], cette ressemblance réside principalement au niveau de la région codant pour le RB, qui a pour caractéristique de se lier spécifiquement à certains récepteurs (ACE2) situés à la surface des cellules infectables [14]. La découverte de ces multiples lignées de coronavirus de pangolin et leur similarité avec le SARS-CoV-2 suggère que les pangolins peuvent être considérés comme des hôtes possibles dans l'émergence du nouveau coronavirus [3].
Pour étudier le rôle des chauves-souris et du pangolin dans la transmission du SARS-CoV-2 à l'homme, Lopes et al (2020) ont effectué une analyse évolutive basée sur les phylogénies moléculaires du virus et de l'hôte de 87 séquences d'acides aminés de la protéine S du CoV. Cette étude a révélé que la diversité des CoVs infectant les chauves-souris, fréquemment associée à une évolution rapide de la propriété de la protéine S des CoVs, pourrait avoir favorisé l'apparition de cette chez l'homme [8].
a- Hypothèses évolutives:
Trois hypothèses ont été avancées par les scientifiques en ce qui concerne l’évolution du génome de SARS-CoV-2:
1- Évolution parallèle: cette hypothèse stipule que des organismes distincts peuvent subir des mutations similaires en réponse à une pression évolutive commune, et donc, développer des traits similaires [9]. Dans le cas du SARS-CoV-2, le domaine RB pourrait avoir été acquis à la suite de mutations qui ont lieu au niveau de cette région, indépendamment du coronavirus du GD pangolin. Pour cette hypothèse, on n’a pas trouvé d’article qui la soutient.
2- Évolution divergente: cette hypothèse suggère que les lignées RaTG13 et Pangolin GD partagent un ancêtre commun, et les différences observées entre ces deux lignées sont dues à une divergence à la suite de substitutions d'acides aminés dans l’une des deux lignées permettant ainsi l’émergence du SARS-CoV-2 [9]. Au même titre que la première hypothèse, on n’a pas trouvé de travaux qui pourraient confirmer cette hypothèse.
3- La recombinaison génétique: c’est l’hypothèse la plus soutenue par les scientifiques [9],[10],[12], elle stipule que des échanges d’informations génétiques ont pu se produire entre différentes lignées de coronavirus par des processus de recombinaisons et de transfères horizontaux de gènes. Selon Lopes et al. (2020), l'occurrence d'une recombinaison entre des CoVs de pangolins et de chauves-souris, ou même une évolution convergente, doit être considérée comme des événements possibles pour l'origine du SARS-CoV-2 [8].
Dans leur étude, Makarenkov et al. (2021) ont tenté de fournir des arguments en faveur de cette dernière hypothèse qui suppose que le génome du SARS-CoV-2 est une chimère des coronavirus RaTG13 et GD Pangolin [9], et aussi, confirmer l’idée que le pangolin est peut-être l’hôte intermédiaire qui a permis au coronavirus de franchir les barrières de l’espèce et devenir pathogène pour l’être humain [9].
Méthodes
Dans leur étude, Makarenkov et al. (2020) ont effectué une analyse approfondie du transfert horizontal de gènes et de la recombinaison, gène par gène, des virus liés au SARS-CoV-2 [9]. Pour cela, plusieurs outils bio-informatique ont été utilisés, parmi eux, le logiciel SimPlot++ dont on va discuter avec plus de détails.
Présentation du logiciel Simplot++
SimPlot++ est un logiciel conçu par Stéphane Samson, Étienne Lord et Vladimir Makarenkov [11], afin de réaliser des analyses de similarité entre séquences génomiques et détecter des recombinaisons de gènes. Ce programme est une version renouvelée de SimPlot (Lole et al., 1999 [7]); plus de fonctionnalités ont été ajoutées, entre autres un outil de contrôle de qualité d’analyse SimPlot et une option multiprocesseur. En plus, beaucoup d’autres modèles de distance pour les graphiques sont disponibles (à présent, il y a vingt modèles de distance d’acides aminés et quarante-trois modèles de distance ADN).
· Création des groupes
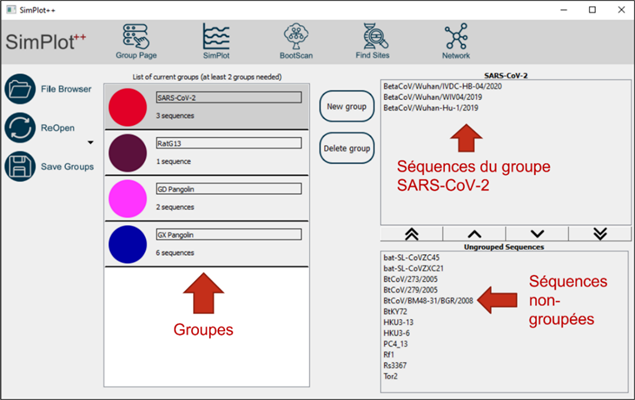
La première étape consiste à importer des séquences alignées et créer des groupes, auxquels des différentes couleurs sont attribuées, comme dans la Figure 1. Un minimum de deux groupes avec une séquence chacun sont exigés [15]. Pour chaque groupe, le programme produit une séquence consensus, qui sera utilisée dans les analyses SimPlot, Bootscan, Network et Recombination.
Figure 1: Création des groupes
· SimPlot et Network
SimPlot (sequence similarity plot) est une représentation graphique du pourcentage de similarité entre un groupe de référence et tous les autres groupes par rapport à la position des nucléotides sur la séquence. La similarité est calculée en utilisant une fenêtre coulissante qui avance pas à pas et compare les caractères des séquences sur un intervalle préétabli, au moyen d’un modèle de distance; donc, il est possible de voir comment la similarité évolue au fil de la séquence. La longueur de la fenêtre (nombre de caractères) et la dimension des pas sont choisies par l’utilisateur, ainsi que le modèle de distance.
Network est l’outil de visualisation de l’analyse SimPlot sous forme de réseau au lieu d'un graphique, et il permet aussi de voir des régions spécifiques du génome. Les nœuds représentent des séquences connectées par des arêtes qui correspondent aux similarités. Celles-ci peuvent être locales (sur des sous-séquences) ou globales (au fil de la séquence entière), les premières en rouge et les dernières en noir. Il faut choisir des seuils de tolérance pour montrer les similarités les plus importantes (par exemple, si on choisit un seuil de 75%, le logiciel ne montrera que les similarités supérieures à 75%).
Figure 2: SimPlot
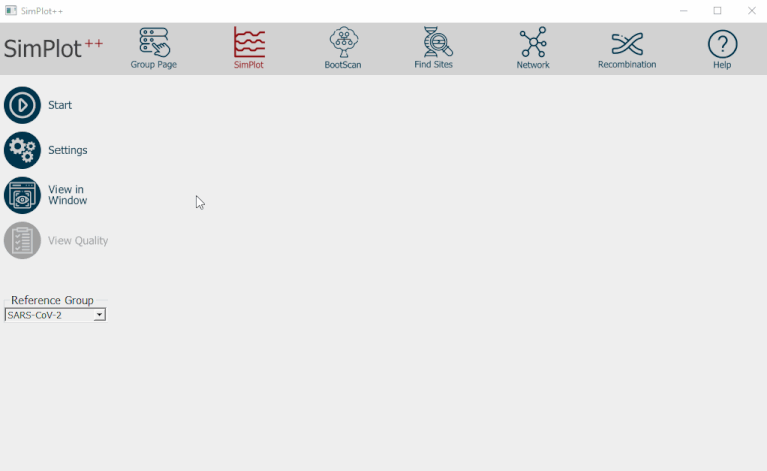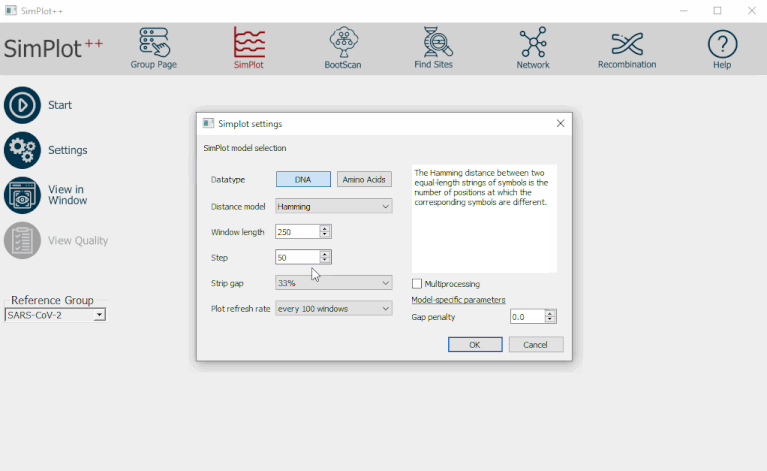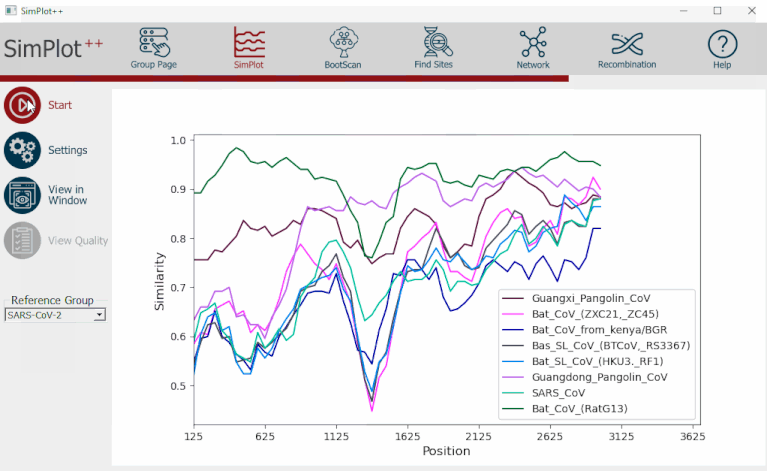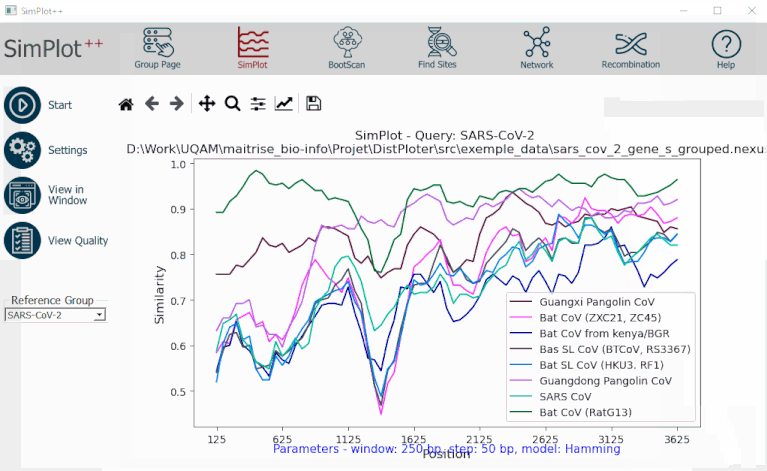
Figure 3: Network
· Bootscan et Find Sites
Les deux analyses Bootscan et Find Sites sont employées pour détecter les régions du génome où des recombinaisons ont eu lieu.
Bootscan se base également sur une fenêtre coulissante pour les calculs; il applique la méthode Bootstrap pour rééchantillonner itérativement les séquences, et pour chaque nouvel alignement multiple obtenu, une matrice de dissimilarités est calculée selon l’un des quarante-trois modèles de distance d’ADN disponibles choisi par l’utilisateur. Ensuite, pour chaque matrice, un arbre phylogénétique est construit par une méthode choisie par l’usager, soit Neighbor-Joining ou UPGMA. Enfin, le pourcentage des arbres dont la séquence est près de la séquence de référence est calculé; donc, dans le graphique, il est possible de voir les groupes qui ont un haut niveau de ressemblance phylogénétique dans différentes positions, comparé au groupe de référence.
Find Sites prend comme données d’entrée quatre séquences et identifie les sites informatifs entre elles, c’est-à-dire, des sites qui ont le même caractère dans deux séquences mais diffèrent dans les autres séquences; cela permet de repérer les possibles recombinaisons. Les séquences à choisir sont: une séquence supposée être générée par des recombinaisons, deux séquences qui pourraient être des possibles parents et une séquence outgroup.
· Recombination
Des tests statistiques peuvent être utilisés pour
détecter des recombinaisons intergéniques et intragéniques, et ainsi valider
l’évidence visuelle obtenue par les graphiques des recombinaisons. Ces tests
sont le ![]() -test,
le NSS, le
-test,
le NSS, le ![]() (dans le PhiPack package
[1]) et le Proportion test. Ce dernier test n’existait pas dans l’ancienne
version de SimPlot, il a été ajouté par Samson et al. (2022).
(dans le PhiPack package
[1]) et le Proportion test. Ce dernier test n’existait pas dans l’ancienne
version de SimPlot, il a été ajouté par Samson et al. (2022).
Le ![]() -test
(PHI, Pairwise Homoplasy Index) a été utilisé par les auteurs pour
investiguer les recombinaisons gène par gène. Il est robuste, simple et il
produit peu de faux positifs par opposition à NSS et
-test
(PHI, Pairwise Homoplasy Index) a été utilisé par les auteurs pour
investiguer les recombinaisons gène par gène. Il est robuste, simple et il
produit peu de faux positifs par opposition à NSS et ![]() [2]. De plus, il peut
être utilisé même s’il y a beaucoup de séquences et de sites, parce qu’il est
efficient du point de vue computationnel, et dans ce scénario, il est aussi
plus puissant [2].
[2]. De plus, il peut
être utilisé même s’il y a beaucoup de séquences et de sites, parce qu’il est
efficient du point de vue computationnel, et dans ce scénario, il est aussi
plus puissant [2].
En testant l’hypothèse nulle d’absence de
recombinaisons, le ![]() -test
donne une valeur p pour chaque gène; si cette dernière est significative, cela
implique la présence d’une recombinaison au niveau de ce gène. En outre, comme
la taille de la fenêtre peut avoir un impact sur les résultats du test, les
auteurs ont utilisé des tailles différentes et ils ont présenté les résultats
avec les valeurs p minimales [9].
-test
donne une valeur p pour chaque gène; si cette dernière est significative, cela
implique la présence d’une recombinaison au niveau de ce gène. En outre, comme
la taille de la fenêtre peut avoir un impact sur les résultats du test, les
auteurs ont utilisé des tailles différentes et ils ont présenté les résultats
avec les valeurs p minimales [9].
Critique de SimPlot++
SimPlot++ représente un bon outil pour analyser la similarité des séquences, puisqu’il offre un éventail de choix, avec une interface moderne et très intuitive aussi pour les non-experts.
L’analyse SimPlot nous donne juste une interface visuelle, donc les résultats qu’on obtient ne sont pas suffisamment concluant, il faut qu’ils soient complétés par les tests statistiques.
Le temps de calcul est raisonnable, et cela grâce à l’option multiprocesseur qui est recommandée surtout dans le cas des grands jeux de données. Cette option permet de faire une analyse de plusieurs fenêtres à la fois.
De plus, l’instrument de contrôle de qualité de SimPlot permet de voir graphiquement s’il y a des erreurs, si le calcul de la distance n’est pas possible dans certaines régions et si le nombre des gaps est trop élevé dans certaines parties.
L’un des inconvénients de SimPlot++ est que dans le cas de l’utilisation d’un grand nombre des groupes la représentation graphique de l’outil n’est pas claire, parce que les courbes se mêlent entre elles et il est difficile de distinguer les groupes.
Une faiblesse potentielle de SimPlot++ qu’on a constaté est l’impossibilité de faire plus d’une analyse à la fois, qui pourrait être utile en termes de temps; en fait, le programme montre une erreur qui indique qu’il y a déjà une analyse en cours.
Résultats
· Analyse phylogénétique des souches de SARS-CoV-2
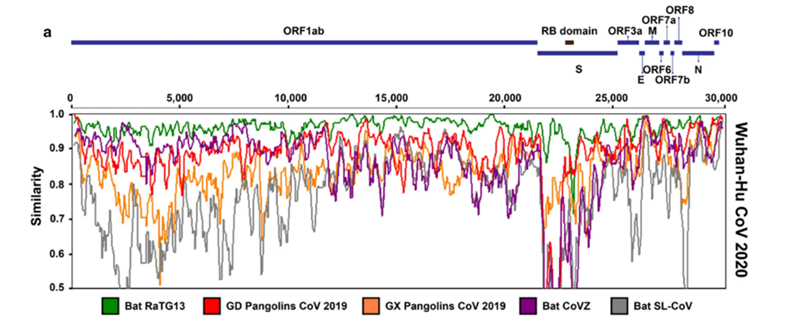
En premier lieu, Makarenkov et al. (2021) ont effectué une analyse de similarité de 25 organismes coronavirus les plus étroitement apparentés au SARS-CoV-2. Cela a été fait à l’aide du logiciel « Simplot++ » qui a permis de comparer le génome de référence du SARS-CoV-2 de Wuhan avec un consensus de génomes de cinq groupes de CoV (CoV RatTG13, les génomes consensus des groupes CoV Pangolin GD et le CoV Pangolin GX, le Bat CoVZ et Bat SL-CoV) [9]. Les groupes sont représentés dans la Figure 4.
Figure 4: Arbre phylogénique des 25 souches de Covs [9]

· Analyse des similarités des génomes SARS-CoV-2
Grace à la fenêtre coulissante « SimPlot++ », il a été possible de visualiser l’évolution des schémas de similarité des séquences entre le génome de référence Wuhan SARS-CoV-2 2020 et les cinq différents groupes, et de détecter ainsi les correspondances entre les similarités de chaque groupe et les régions codantes du génome du SARS-CoV-2. Les résultats obtenus avec cette analyse confirment les résultats obtenus avec d’autre études [8], [10], [12], à savoir que le SARS-CoV-2 de Wuhan et du RaTG13 partagent 96 % d'identité au niveau du génome entier, tandis que les génomes du SARS-CoV-2 de Wuhan et du Pangolin GD sont identiques à 90 %. Les génomes du CoV de RaTG13 et de GD Pangolin sont donc de loin les plus proches du génome du SARS-CoV-2 [9], [10].
Figure 5: Analyse SimPlot du génome entier [9]
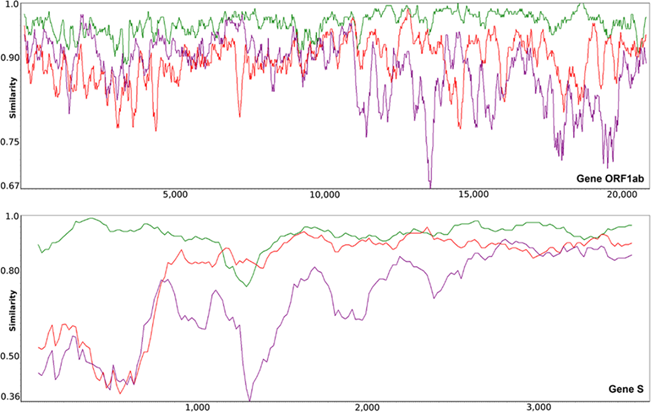
· Analyse des similarités des gènes ORF1ab et S de SARS-CoV-2
Dans l’étude de Makarenkov et al. (2021), une analyse détaillée des similitudes au niveau des gènes individuels entre les séquences génétiques du SARS-CoV-2 de Wuhan et les séquences du groupe RaTG13, du groupe CoV de GD Pangolin et du groupe Bat CoVZ a été effectuée. L’analyse par fenêtre coulissante qu’ils ont réalisée a permis de traiter chaque segment génétique considéré comme une région génétique indépendante ayant sa propre histoire évolutive [9]. Cette analyse (gène par gène) a révélé que certaines régions du SARS-CoV-2 étaient plus similaire au CoV du Pangolin GD qu'au RaTG13. Ces régions correspondaient au gènes S (au niveau du domaine RB), ORF3a, M, ORF7a et N. Cela les a laissés suggérer que les événements de recombinaison entre le CoV GD Pangolin et RaTG13 n’ont pas eu lieu seulement au niveau du gènes S, mais aussi dans quatre autres gènes de ce génome de CoV [9]. Ils ont même constaté que dans certaines régions génétiques continues, en particulier dans les gènes ORF1ab, ORF3a, M et N, les séquences génétiques du SARS-CoV-2 sont beaucoup plus similaires à celles des virus CoV ZC45 et ZXC21 de la chauve-souris qu'à celles des CoV du pangolin GD, et parfois même de RaTG13 [9].
Figure 6: Analyse SimPlot du gène ORF1ab et du gène S
Afin de détecter et de valider ces événements de transfert de gènes et de recombinaison, trois méthodes différentes ont été utilisés par Makarenkov et al. (2021): analyse de recombinaison Φ-test et analyse HGT intergénique et intra-génique. Les résultats obtenus par ces trois approches ont confirmé encore une fois que des transferts de gène entre le Guangdong Pangolin CoV et le SARS-CoV-2 ont bien eu lieu au niveau du gène S.
Discussion
La protéine S du CoV est une protéine d'enveloppe qui joue le rôle le plus important dans l'attachement, la fusion et l'entrée du virus dans les cellules hôtes [10]. Une telle similitude de structure et de séquence au niveau de cette protéine, indique fortement une évolution convergente entre les RBD du SARS-CoV-2 et du SARS-CoV pour une meilleure liaison à l'ACE2 [5]. Ainsi, il est considéré que l'interaction ACE2 est la barrière d'hôte qui protégeait les humains des agents pathogènes zoonotiques. Le SARS-CoV-2 peut avoir nécessité donc, certains hôtes mammifères intermédiaires pour passer aux humains et déclencher l'épidémie [8].
Zhu et al. (2020) ont trouvé une séquence de 228 pb de long dans la protéine S du SARS-CoV-2 qui est probablement une séquence intégrée résultant d'une recombinaison entre certaines souches similaires au Bat-CoV-RaTG13 et certaines souches similaires au Pangolin-CoV-2019. Zhu et al. (2020) ont aussi confirmé qu’il y avait une forte probabilité que le SARS-CoV-2 provient d'un coronavirus de chauve-souris après intégration par recombinaison d'un fragment d'ARN d'un coronavirus de pangolin dans le gène de la protéine S. Plusieurs analyses phylogénétiques basées sur la protéine S de SARS-CoV-2 soutient l'hypothèse selon laquelle la chaîne de transmission de SARS-CoV-2 est partie de la chauve-souris, a eu le pangolin malais comme hôte intermédiaire et a infecté les humains [9], [8], [10], [12].
Makarenkov et al. (2020) ont également effectué une analyse en grappes des arbres génétiques des coronavirus afin d'identifier les gènes ayant une histoire évolutive similaire. Cette analyse a révélé la présence de trois groupes de phylogénies de gènes possibles. L’identification de ces trois groupes a permis non seulement d'identifier les gènes et les génomes qui ont été affectés par la recombinaison, mais aussi de déterminer les organismes donneurs et receveurs pour chaque événement de recombinaison détecté, et de savoir si cet événement était intra-génique ou intergénique [9].
Application
· Analyse de similarité des génomes du SARS 2003
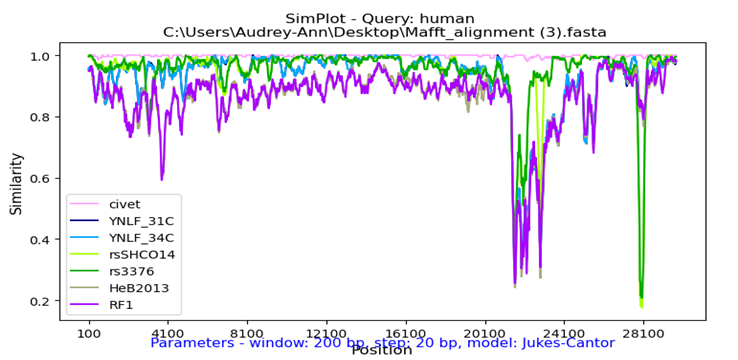
Nous avons tenté une analyse en parallèle d’un jeu de données de souches de SARS-CoV-1, dans l’optique de voir si le logiciel permettrait de détecter d’autres types de transferts génétiques, décrits par la littérature, qui auraient mené à l’évolution du virus vers une souche capable d’infecter l’humain.
Dans le cas du SARS-CoV-1, la civette a été décrite comme l’hôte intermédiaire ayant mené au Sars humains. Les chauves-souris sont, encore une fois, le réservoir naturel de ces souches pathogènes. Les données de phylogénie nous indiquent que ce coronavirus aurait été transmis de l’espèce Rhinolophus Sinicus (rs3376, rsCHSO14) vers la civette, à la suite d’un événement de transfert horizontal du gène ORF8 avec une espèce de Rhinolophus Ferrumequinum (YNFL_31C, YNLF_24C) [6].
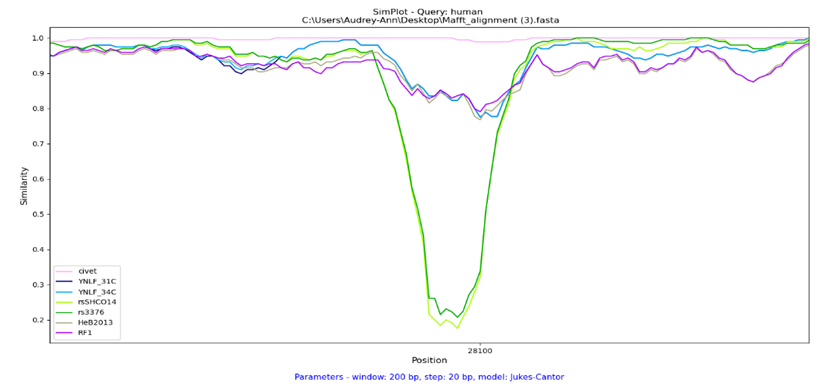
L’analyse SimPlot par rapport au Sars humain montre effectivement une plus grande similitude globale du génome des souches R. Sinicus par comparaison aux autres souches de chauve-souris. Tous comme chez SARS-CoV-2, on observe une large variation au niveau du gène S. Cette région reste toutefois largement similaire à la souche de la civette, et montre la capacité de celle-ci d’être transmise de façon interchangeable entre les deux espèces. Précisément à la position du gène ORF8 (27 000 - 28 000), on observe une grande baisse de similitude des virus ancestraux rs3376 et rsCHSO14, par opposition à une similitude plus marquée pour les deux espèces YNFL. L’analyse appuie donc la théorie du transfert de gène de R. Ferrumequinium vers la descendance des espèces S. Sinicus ayant mené aux Sars adaptés à la transmission entre humains et civettes.
Figure 7: Analyse SimPlot du génome de SARS-CoV-1 et du gène ORF8
Conclusion
Les conclusions auxquelles Makarenkov et al. (2021) ont abouti par cette étude est que les événements de recombinaison confirmés dans les gènes S et N ainsi que l'analyse de recombinaison « SimPlot++ » des gènes S, ORF3a, M, ORF 7a et N suggèrent que le pangolin du Guangdong est un hôte intermédiaire probable du SARS-CoV-2 [9], et c’est lui qui a permis sa transmission de la chauve-souris à l’homme. Leurs résultats révèlent aussi que l’émergence du SARS-Cov-2 n’est pas dû seulement à une recombinaison et un transfert de gène entre les CoVs RatTG13 et Pangolin GD, mais aussi entre les coronavirus CoVZC45 et CoVZXC21 de la chauve-souris ou leur ancêtre commun [9].
Références
Références bibliographiques:
[1] Bruen, T., & Bruen, T. (2005). PhiPack: PHI test and other tests of recombination. McGill University, Montreal, Quebec. PhiPack-PHI-test-and-other-tests-of-recombination.pdf (researchgate.net)
[2] Bruen, T. C., Philippe, H., & Bryant, D. (2006). A simple and robust statistical test for detecting the presence of recombination. Genetics, 172(4), 2665-2681.
https://doi.org/10.1534/genetics.105.048975
[3] Guo, YR., Cao, QD., Hong, ZS. et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak – an update on the status. Military Med Res 7, 11 (2020). https://doi.org/10.1186/s40779-020-00240-0.
[4] Holmes, EC., Goldstein, SA., Rasmussen, AL. et al. A critical review. Cell. 2021 Sep 16;184(19):4848-4856. https://pubmed.ncbi.nlm.nih.gov/34480864/.
[5] Lan, J., Ge, J., Yu, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020). https://doi.org/10.1038/s41586-020-2180-5.
[6] Lau, S. K., Feng, Y., Chen, H., Luk, H. K., Yang, W. H., Li, K. S., ... & Woo, P. C. (2015). Severe acute respiratory syndrome (SARS) coronavirus ORF8 protein is acquired from SARS-related coronavirus from greater horseshoe bats through recombination. Journal of virology, 89(20), 10532-10547. https://doi.org/10.1128/JVI.01048-15
[7] Lole, K. S., Bollinger, R. C., Paranjape, R. S., Gadkari, D., Kulkarni, S. S., Novak, N. G., ... & Ray, S. C. (1999). Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. Journal of virology, 73(1), 152-160. https://doi.org/10.1128/JVI.73.1.152-160.1999
[8] Lopes LR, de Mattos Cardillo G, Paiva PB. Molecular evolution and phylogenetic analysis of SARS-CoV-2 and hosts ACE2 protein suggest Malayan pangolin as intermediary host. Braz J Microbiol. 2020 Dec;51(4):1593-1599. https://pubmed.ncbi.nlm.nih.gov/32592038/.
[9] Makarenkov, V., Mazoure, B., Rabusseau, G. et al. Horizontal gene transfer and recombination analysis of SARS-CoV-2 genes helps discover its close relatives and shed light on its origin. BMC Ecol Evo 21, 5 (2021). https://doi.org/10.1186/s12862-020-01732-2.
[10] Malaiyan J, Arumugam S, Mohan K, Gomathi Radhakrishnan G. An update on the origin of SARS-CoV-2: Despite closest identity, bat (RaTG13) and pangolin derived coronaviruses varied in the critical binding site and O-linked glycan residues. J Med Virol. 2021 Jan;93(1):499-505. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7361880/
[11] Samson, S., Lord, É., & Makarenkov, V. SimPlot++: a Python application for representing sequence similarity and detecting recombination. arXiv preprint arXiv:2112.09755 (2021). https://doi.org/10.48550/arXiv.2112.09755
[12] Zhu, Z., Meng, K. & Meng, G. Genomic recombination events may reveal the evolution of coronavirus and the origin of SARS-CoV-2. Sci Rep 10, 21617 (2020). https://doi.org/10.1038/s41598-020-78703-6.
[13] Zhukova, A., Blassel, L., Lemoine, F., Morel, M., Voznica, J., & Gascuel, O. (2021). Origin, evolution and global spread of SARS-CoV-2. Comptes Rendus. Biologies, 344(1), 57-75. https://doi.org/10.5802/crbiol.29
Références internet:
[14] Mouscaz Y. Analyses bioinformatiques du coronavirus 2019-nCoV : pourquoi et comment? (2020). https://bioinfo-fr.net/analyses-bioinformatiques-du-coronavirus-2019-ncov-pourquoi-et-comment.
[15] Samson, S. Simplot+
https://github.com/Stephane-S/Simplot_PlusPlus/blob/master/README.md#SimPlot-analysis