Détection
et annotation de régions fonctionnelles non codantes du génome humain
Présentée par Mathieu Blanchette
Rapport de conférence par
Christian Poitras et Maxime Benoît-Gagné
Table des matières
5.2 Score de parcimonie attendu_ 4
5.3 Score de parcimonie observé_ 6
7.2 Calculs et logiciels utilisés
1
Introduction
Les régions
fonctionnelles non codantes du génome humain contiennent des éléments de
régulation de la transcription et de l’épissage, des gènes à ARN (acide
ribonucléique) et des régions permettant l’attachement des chromosomes à la
matrice. La régulation de la
transcription permet de réguler quels gènes seront transcrits et en quelle
quantité. La régulation de l’épissage
permet l’épissage alternatif, c’est-à-dire le choix de la ou des protéine(s)
qui seront exprimée(s) à partir d’un seul gène.
Toutes les cellules
somatiques d’un même individu partagent le même matériel génétique. Ainsi, les régions génomiques fonctionnelles
non codantes sont celles qui gèrent l’expression différentielle des protéines
entre les cellules différentes d’un même individu.
On estime que ces
régions représenteraient 3,5% du génome humain. Un autre 1,5% du génome
serait constitué de gènes codant des protéines. Ce qui donne un total de 5% du génome formé
par des régions fonctionnelles.
La recherche menée
par Dr Mathieu Blanchette et ses collègues s’insère dans ce domaine. Le but étant de trouver où sont ces régions
fonctionnelles non codantes et quels sont leurs rôles.
2
Conservé donc
fonctionnel
L’hypothèse sur
laquelle se fonde tous les travaux faits par cette équipe est qu’une mutation
affecte différemment la probabilité de survie et de reproduction d’un individu
selon que celle-ci survienne dans une région fonctionnelle ou non
fonctionnelle. Une mutation dans une
région fonctionnelle modifie l’individu le plus souvent en diminuant ses
chances de survie et de reproduction.
Alors qu’une mutation dans une région non fonctionnelle ne modifie pas
le phénotype de l’individu. Ses chances
de survie et de reproduction sont conservées.
Une mutation chez un
individu qui peut se reproduire et donc transmettre son matériel génétique est
conservée plus facilement qu’une mutation chez un individu qui ne se reproduira
pas et ne transmettra donc pas son matériel génétique. C’est pourquoi les régions fonctionnelles
accumulent des mutations à un taux moindre que les régions non
fonctionnelles. Ce phénomène est bien
illustré en traçant deux graphiques de la fréquence en fonction du degré de
conservation pour des régions du génome, un graphique pour des régions
probablement non fonctionnelles (les régions répétées) et un autre pour des
régions couvrant tout le génome.
La courbe du
graphique pour les régions non fonctionnelles est déplacée vers la gauche par
rapport à celle du graphique pour l’ensemble des régions. Cela indique que les régions non
fonctionnelles sont moins conservées.
3
Exemple de
conservation
Passons à un exemple
célèbre pour démontrer que les régions fonctionnelles sont souvent
conservées. L’ARN ribosomal est une
région très importante du génome. Il
code pour des composantes essentielles des ribosomes. Même chez des espèces divergentes, cet ARN ribosomal est
remarquablement bien conservé (1).
4
Projet ENCODE
Maintenant que le
génome humain est séquencé, il devient intéressant d’interpréter les
séquences. Le projet ENCODE concentre
les efforts de séquençage et d’annotation sur un groupe de loci représentant 1%
du génome humain. Dr Blanchette et ses
collègues se sont attardés sur la région autour du gène CFTR (le gène muté dans
la fibrose kystique du pancréas) faisant partie de ce 1%.
Ils y ont cherché
les séquences conservées entre 13 vertébrés dont l’homo sapiens. Ils ont utilisé les séquences souvent
incomplètes de cette région des organismes étudiés. Ils ont utilisé un arbre phylogénétique dont les branches sont
valués par les taux de mutations neutres.
5
Algorithmes
5.1
Score de
parcimonie
Nous avons besoin
d’une façon de calculer si des régions spécifiques sont conservées entre les
différentes espèces étudiées. Pour le faire, nous allons utiliser les scores de
parcimonie.
Un score de
parcimonie est un score minimal représentant les changements biologiques au
travers d’une spéciation comme, par exemple, une arête dans un arbre de
phylogénie. Lorsque nous avons un arbre de phylogénie, il est possible de
calculer un score qui représente les changements les plus probables (qui ont un
coût faible) qui se sont produits pendant l’évolution. Ce score peut être
calculé en n’ayant comme informations que les caractéristiques des espèces
observables aujourd’hui. Les caractéristiques seront les nucléotides de la
région CFTR. Il faut, de plus, que l’arbre phylogénique donne de l’information
sur la distance entre les espèces. La distance représente la probabilité qu’un
nucléotide se transforme en un autre lorsqu’il s’est produit une spéciation
entre deux espèces ayant un ancêtre commun.
Voici un exemple où
nous allons vérifier le nombre minimal de mutations qui peuvent mener à
l’évolution du nucléotide en rouge.
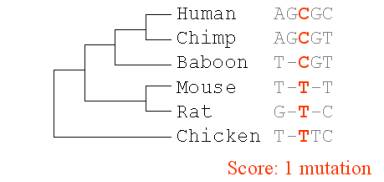
Nous pouvons
constater que l’évolution minimale donnant ces nucléotides a commencé avec un
nucléotide T qui a muté vers un C chez l’ancêtre du babouin.
Voici un score de
parcimonie de 3 mutations.
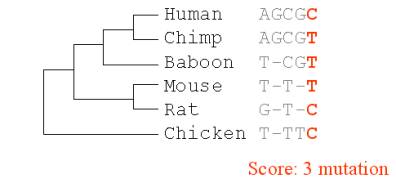
Nous pouvons
constater que le nucléotide est plus conservé dans l’exemple avec une mutation
que dans le dernier exemple, où le nombre de mutations nécessaires est plus
élevé. Ceci indique que l’évolution est plus aléatoire dans le dernier cas et
que la structure est probablement mieux conservée dans le cas où il n’y a
qu’une seule mutation.
Notons également que
s’il nous manque de l’information sur une espèce à un certain nucléotide, il ne
sera pas considéré dans le score de parcimonie. Les trous (gaps) dans une
séquence ne seront pas considérés. Voici un exemple, où le score de parcimonie
est probablement inférieur à la réalité.
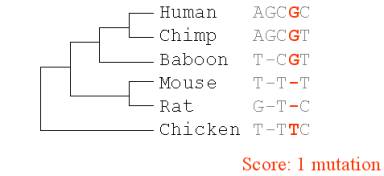
5.2
Score de
parcimonie attendu
Pour savoir si une
région est conservée entre plusieurs espèces, nous devons savoir quel est le
score de parcimonie auquel nous nous attendons si l’évolution a modifié des
nucléotides d’une façon aléatoire, soit une évolution neutre. En calculant la
valeur pour une évolution neutre, il nous sera possible de comparer cette
valeur avec un score de parcimonie trouvé pour les espèces étudiées.
Pour calculer le
score de parcimonie attendu, nous pouvons utiliser des formules mathématiques à
partir d’un arbre donné ou nous pouvons faire des essais aléatoires de
mutations qui nous donnerons un score de parcimonie attendu.
Voici un arbre
d’évolution qui nous donne, sur chaque arête, la probabilité qu’un nucléotide
soit modifié. Notons que la probabilité qu’un nucléotide soit modifié est
généralement dépendante de la distance séparant deux espèces.
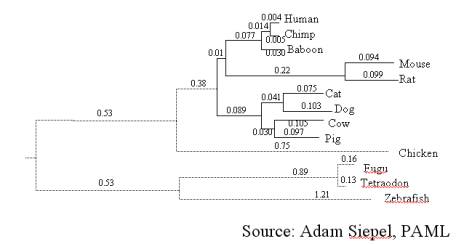
À partir de cet
arbre, nous pouvons faire des essais aléatoires de mutations pour un
nucléotide. Voici un exemple d’une évolution possible qui tient en compte les
probabilités qu’un nucléotide soit modifié à chaque spéciation.
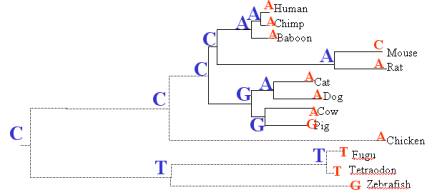
Dans cet exemple,
nous constatons que le nucléotide a muté 9 fois. Le score de parcimonie, étant
une borne inférieure expliquant comment l’évolution s’est produite, sera, en
réalité, plus bas que 9. Comme nous ne devrions pas avoir d’information sur les
espèces ancestrales, nous devons tenir compte que des espèces qui sont situés
dans les feuilles. Nous devons ensuite reconstruire l’arbre avec un minimum de
mutations en ne tenant compte que des feuilles.
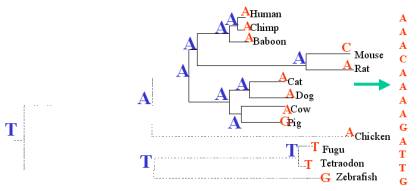
Cet arbre nous
montre que le score de parcimonie tiré de l’arbre aléatoire précédent n’est pas
de 9 mutations comme nous l’aurions souhaité, mais de 4 mutations. Il a été
possible de reconstruire l’arbre avec seulement 4 mutations. C’est ce genre de
score de parcimonie que nous allons utiliser pour savoir à quel score de
parcimonie nous devrions nous attendre.
En faisant plusieurs
essais, nous obtenons une courbe indiquant le nombre de mutations auquel nous
devrions nous attendre dans une évolution neutre.
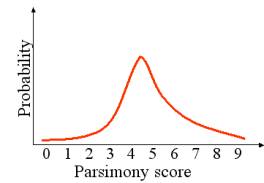
Nous constatons
qu’il est normal d’obtenir un score de parcimonie de 4 ou plus. Un score de
parcimonie de 2 serait intéressant pour un nucléotide, mais il faut aussi
considérer que nous obtiendrons quelques fois, par hasard, des scores de
parcimonie inférieurs à 4 même pour une évolution neutre.
Ajoutons qu’il est
intéressant d’inclure dans les calculs aléatoires un coût pour transformer un
certain nucléotide en un certain autre. Ces coûts peuvent être stockés dans une
matrice de coûts et peuvent affecter la façon dont les nucléotides changent
modifiant ainsi la courbe des scores de parcimonie attendus.
5.3
Score de
parcimonie observé
Après avoir obtenu
une courbe des scores de parcimonie attendus, nous devons calculer les scores
de parcimonie observés pour chaque nucléotide. Nous allons les calculer de la
même façon que dans l’exemple donné plus haut.
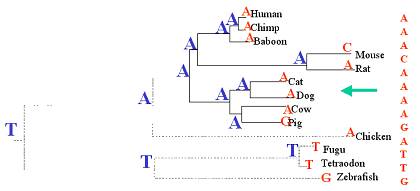
Si les espèces ont
les nucléotides tels que donnés dans l’exemple, nous obtenons un score de
parcimonie de 4. Notons que nous devons connaître l’arbre d’évolution pour calculer
le score de parcimonie attendu. Il n’est pas nécessaire de connaître les
probabilités qu’un nucléotide change pour chaque arête pour calculer un score
de parcimonie observé, car nous ne calculons que le nombre de mutation. Nous
devons connaître les probabilités seulement pour faire le calcul des scores de
parcimonie attendus.
5.4
Surprise
Une fois que les
scores de parcimonie observés et attendus sont calculés, nous devons également
vérifier si des régions de l’ADN sont conservées. Pour ce faire, nous allons
calculer une surprise. La surprise est une représentation numérique indiquant
si nous devons considérer une région de l’ADN comme étant conservée. La
surprise ne garantit pas que la région est réellement conservée, car elle peut,
dans certain cas, être due au hasard. Voici un exemple où nous considérons la
probabilité d’obtenir un certain score de parcimonie.
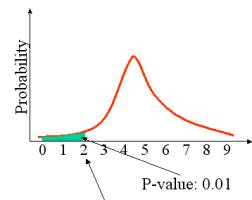
Le score de
parcimonie observé ici est 2. Le p-value est de 0,01. Le p-value est la
probabilité d’obtenir cette valeur de score de parcimonie ou un score
inférieur. Le p-value est calculé selon l’aire sous la courbe allant de 0
jusqu’au score de parcimonie observé. Le p-value est une bonne façon de
représenter la surprise. Plus le p-value est élevé, plus la probabilité d’obtenir
cette valeur de score de parcimonie, ou une valeur inférieure, par hasard est
élevée.
Notons qu’il est
également nécessaire d’avoir plusieurs espèces pour avoir une courbe de
parcimonie qui permet d’avoir une bonne surprise. Si les espèces sont trop
proche l’une de l’autre, il sera difficile d’avoir une surprise élevée (un
p-value bas).
Voici un exemple où
le score de parcimonie ne sera pas très utile si l’on considère les mutations
au niveau de l’ADN seulement.
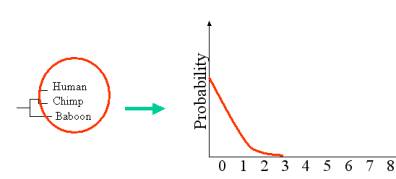
Même avec un score
de parcimonie de 0 ou 1, le p-value sera très élevé. C’est pour cette raison
que nous devons considérer plusieurs espèces assez éloignées.
5.5
Fenêtre
coulissante
La surprise ne peut
pas être calculée sur un seul nucléotide, alors nous la
calculons sur 25 nucléotides. Ceci nous permet d’éviter d’avoir une
surprise élevée due au hasard, mais nous pouvons perdre certaines informations
comme une petite séquence très conservée qui a une utilité importante mais qui
est entourée de régions ayant une surprise très faible.
Voici la formule
utilisée pour calculer la surprise sur 25 nucléotides.
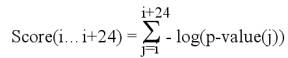
En prenant des
séquences de nucléotides, nous pouvons observer les régions conservées. Voici
un exemple de régions conservées pour le gène CORTBP2.

Lorsque ParsPVal est
élevé, il indique une bonne surprise. On peut constater que plusieurs régions
sont conservées à l’extérieur des régions codantes (indiquées par les blocs
sous RefSeq Genes). Il serait intéressant de faire des expériences sur ces
régions pour savoir si elles ont une utilité.
5.6
Implémentation
Un programme
permettant de calculer les scores de parcimonie est disponible. Ce programme se
nomme «FootPrinter» et il a été programmé par Mathieu Blanchette. Nous avons
utilisé ce programme, avec un second programme qui permet de calculer les
scores de parcimonie selon les fenêtres coulissantes, pour obtenir les
résultats présentés plus loin.
Nous avons expliqué
le principe des scores de parcimonie pour obtenir un score pour un seul
nucléotide à la fois. Notons qu’il est plus performant de calculer des scores
de parcimonie en traitant des chaînes de caractères plutôt que des nucléotides
simples. Cette méthode de calcul est utilisée dans le programme «FootPrinter».
Les distances entre les chaînes sont calculées avec la distance de Hamming qui
représente le nombre minimum de substitutions pour qu’une chaîne soit
transformée en une autre.
Pour ceux qui
veulent utiliser le programme «FootPrinter», il faut fournir en paramètre un
arbre de phylogénie où les distances sont la probabilité qu’un nucléotide
change d’un nœud à un autre dans l’arbre et des séquences de nucléotides dans
un format FASTA.
L’algorithme possède
une complexité O(n*min(l*k*(3k)d/2, k*(4k+l)))
où n est le nombre de taxa, l est la longueur de la séquence
complète à traitée, k est la longueur d’une chaîne traitée (une partie
de la séquence) et d est la distance maximale que l’on peut atteindre
avant de laisser tomber cette séquence.
Pour ceux qui
voudraient savoir comment l’algorithme, implémenté dans le programme
«FootPrinter», fonctionne en détail, je suggère de lire le document «Algorithms
for phylogenetic footprinting» que l’on peut trouver sur le site Internet de M.
Mathieu Blanchette en suivant le lien suivant : http://www.cs.washington.edu/homes/blanchem/papers.html.
De plus, le document contient les temps d’exécution du programme sur un
ordinateur dans différentes situations.
Le programme
«FootPrinter» qui exécute le calcul des scores de parcimonie se trouve sur le
site http://bio.cs.washington.edu/software.html.
6
Résultats
Dr Blanchette et ses
collègues ont trouvé des régions conservées parmi des exons connus et des
régions non codantes. La figure 1
représente la proportion des régions conservées selon leur localisation.
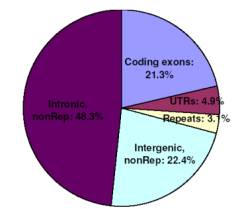
Figure 1: Diagramme circulaire représentant la proportion des régions conservées du grand CFTR selon leur localisation. Intronic nonRep : régions introniques non répétitives, Intergenic nonRep : Régions intergéniques non répétitives, Coding exons : exons codants, UTRs : régions non traduites et Repeats : répétitions.
6.1
Exons
La conservation
inter-espèce au niveau des exons est habituellement nette. La figure 2a le montre.
La figure 2b montre
la conservation inter-espèce au niveau d’un gène soumis à un mécanisme
d’épissage alternatif. Autour de l’exon
étant la cible de l’épissage alternatif (parfois présent dans l’ARN messager et
dans la protéine et parfois absent), la région génomique est bien
conservée. Ce qui mène à l’hypothèse
que cette région est fonctionnelle et que sa fonction est la régulation de
l’épissage alternatif de ce gène.
a
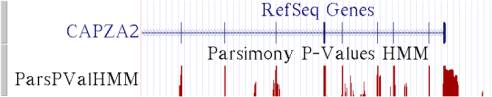
b

Figure 2. Conservation inter-espèce au niveau de deux gènes. En haut de chaque graphique, on voit les exons (bandes verticales bleues). En bas, on voit des mesures de la conservation (pics rouges). RefSeqGenes : gènes bien annotés selon le NCBI. ParsPValHMM : valeurs-p de parcimonie avec un modèle de Markov caché. (a) Gène CAPZA2. (b) Gène ST7.
6.2
Régions non
codantes
Passons aux régions non
codantes mais conservées. Comme
l’indique la figure 1, certaines furent détectées.
Certaines régions
conservées contiennent un groupe de possibles sites de liaison de facteurs de
transcription. La figure 3 donne
l’exemple d’un intron du gène MET. Les
sites de liaison de facteurs de transcription sont importants pour la
régulation de la transcription.
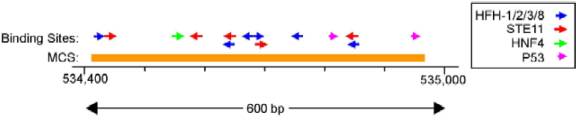
Figure 3. Une région de 600-pb
située à l’intérieur d’un intron du gène MET.
Cette région contient un groupe de possibles sites de liaison pour les
facteurs de transcription indiqués. La
barre orange délimite la position d’une séquence conservée entre plusieurs
espèces. Noter que cette séquence
conservée entre plusieurs espèces est flanquée d’une séquence intronique de 4,6
kb en amont et de 26 kb en aval. Deux
des sites de liaison au facteur de transcription HFH (hepatocyte nuclear
factor homolog : homologue du facteur nucléaire
hépatocytaire) se superposent. Il n’y a
donc que six occurrences indépendantes de ce facteur de transcription. Binding Sites : sites de liaison,
MCS(« Multi-Species Conserved Sequence ») : séquences conservées
entre plusieurs espèces, bp : paires de bases et p53 (un
anti-oncogène(4)).
D’autres régions
conservées concernent des éléments structuraux d’ARN. Le logiciel QRNA a été utilisé sur chaque région conservée. 29 de ces 904 régions ont une structure
secondaire d’ARN conservée.
12 de ces 29 régions
sont localisées dans une région non traduite 3’ ou 5’. La région non traduite 5’ (5’ UTR) est la
partie de l’ARN messager en amont du codon d’initiation de la traduction. La région non traduite 3’ (3’ UTR) est la
partie de l’ARN messager en aval du codon de terminaison de la traduction. La figure 4 illustre l’exemple de la
conservation inter-espèce de la région non traduite 3’ du gène CAV1. Cet exemple montre la possibilité de
l’existence d’une fonction pour cette région non codante. Cet exemple montre la possibilité de l’existence
d’une fonction pour cette région non codante.
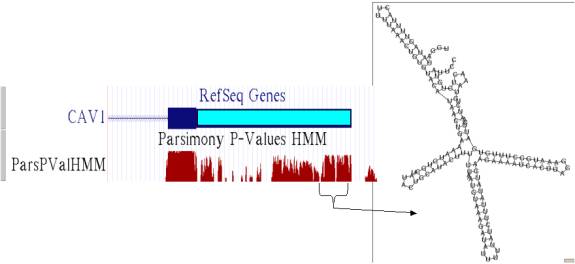
Figure 4. Mesure de la
conservation de la région 3’ non traduite du gène CAV1 et illustration de la
structure secondaire d’ARN de la région indiquée par une accolade. Région traduite (bleu), région de l’ARN
messager non traduite (turquoise), adénine (A), cytosine (C), guanine (G) et
uracile (U).
10 des 29 régions
conservées inter-espèces possédant possiblement des éléments structuraux d’ARN
sont situées dans des introns. Les
introns sont les parties du gène éliminées de l’ARN mature (4). Dr Blanchette et ses collègues ont posé la
condition supplémentaire suivante aux séquences dites introniques. La région intronique doit être à moins de 2
kb d’un exon. La figure 5 donne
l’exemple d’une séquence intronique conservée dans un intron du gène ST7. Les nucléotides de cette région sont
parfaitement conservés sauf un au bout d’une épingle à cheveux (hairpin). Cette conservation suggère une fonction
importante peut-être liée à l’épissage.
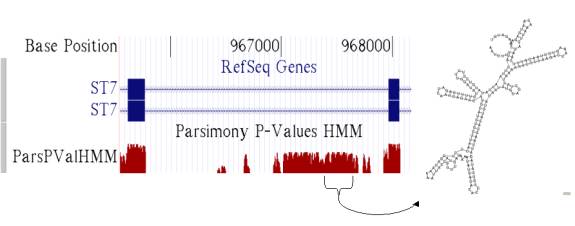
Figure 5. Mesure de la
conservation pour une séquence intronique située dans un intron du gène
ST7. Une illustration de la structure
secondaire d’ARN de la région indiquée par une accolade est donnée. Base position : position du nucléotide.
Des régions
introniques contenant des éléments structuraux d’ARN loin des exons ont aussi
été identifiées. La figure 6 présente
l’exemple d’une telle séquence située en plein milieu des exons 1 et 2 du gène
ST7.
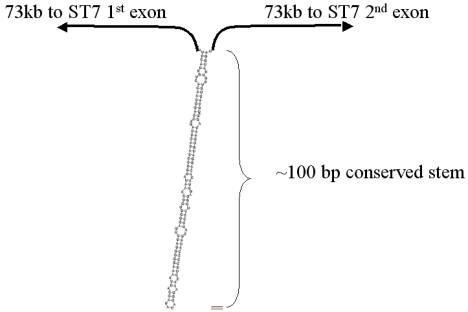
Figure 6. Illustration de la
structure secondaire d’ARN d’une région conservée entre plusieurs espèces. Cette région est au milieu exact des introns
1 et 2 du gène ST7. 100 bp conserved
stem : tige conservée d’environ 100 pb, 1st : premier et 2nd :
deuxième.
7
Notre expérience
7.1
Introduction et données
Nous avons appliqué
la même méthode à une autre séquence.
Il s’agit de la séquence du gène codant le précurseur de la protéine 29
du reticulum endoplasmique (aussi appelée protéine luminale du reticulum
endoplasmique Erp28). Le nom de ce gène
dans le NCBI est C12orf18. Nous avons
utilisé une longue séquence en 5’ du gène et une courte séquence en 3’ du
gène. La longueur totale est 11,672 kb.
Des séquences
orthologues sont connues au moins pour la souris et le rat (8). Nous avons utilisé les séquences sur les
génomes de l’humain, de la souris et du rat sur le NCBI. En comparant ces trois séquences, nous avons
cherché des régions conservées. L’arbre
utilisé par le logiciel «FootPrinter» était le même que celui de la conférence
que nous avons élagué des branches inutiles.
Il est illustré à la figure 7.
Nous avons aligné
ces séquences par ClustalW.
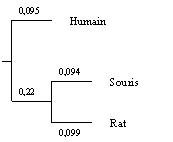
Figure 7. Arbre phylogénétique ayant servi de modèle
d’évolution neutre.
7.2
Calculs et
logiciels utilisés
Pour obtenir les
résultats, nous avons utilisé plusieurs logiciels dont certains sont faits
maison. Pour faire l’alignement des différentes espèces, nous avons utilisé
ClustalW. Nous devions transformer les résultats de ClustalW en format FASTA
pour l’utiliser dans le logiciel «FootPrinter». Nous avons créé un programme
maison capable de transformer des données venant de ClustalW en format FASTA.
Ensuite, nous avons utilisé «FootPrinter» pour obtenir les score de parcimonie
et de p-value des différents acides nucléiques. Les p-value étant calculés pour
un nucléotide à la fois, nous avons créé un logiciel qui permet d’obtenir les
scores de p-value pour des fenêtres de 25 nucléotides selon la formule
suivante.
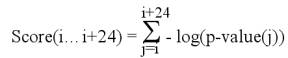
Pour simplifier le
traitement, ce programme donne des résultas dans un format texte délimité. Les
résultats pouvaient être importés directement dans le logiciel «Excel». Nous
avons utilisé «Excel» pour obtenir le graphique montré plus bas. Notons que la
partie du graphique montrant la position des exons a été obtenue par un second
programme donnant un fichier texte délimité dont les résultats ont aussi été
importés dans «Excel». Ce programme prenait l’alignement tiré de ClustalW et
calculait la position réelle des exons en tenant compte des gaps ajoutés. Nous
avons trouvé la position des exons ainsi que leur taille sans gaps sur le site
du NCBI.
7.3
Nos résultats
Finalement, nous
avons obtenu la surprise (ou degré probable de conservation) pour chaque
position de la séquence humaine alignée (donc contenant des gaps). Un gap est considéré comme occupant une
position au même titre qu’un nucléotide.
Un logiciel maison a
permis de trouver des positions particulières sur la séquence avec gaps en
connaissant la longueur des sous-séquences correspondantes. Les sous-séquences en question énumérées du
5’ vers le 3’ sont: la région proximale 5’, l’exon 1, l’intron 1, l’exon 2,
l’intron 2, l’exon 3, la région non-traduite 3’ et la région distale 3’. Nous connaissons donc les positions limites
entre ces régions de la séquence génomique.
Cela nous a permis de réaliser la figure 8.

Figure 8. Lien entre la conservation inter-espèces et
la localisation des exons pour la région englobant le gène C12orf18 chez
l’humain. (En haut.) Localisation des exons sur la séquence
contenant des gaps. Les exons sont
disposés de 5’ vers 3’ c’est-à-dire exon 1, exon 2 et exon 3. (En bas.)
Degré probable de conservation en fonction de la position du nucléotide
sur la séquence avec des gaps. La
séquence est représentée du 5’ vers le 3’.
7.4
Interprétation
Le résultat le plus
évident en observant la figure 8 est la conservation des exons 2 et 3. Nous ne savons pas pourquoi l’exon 1 n’est
pas conservé alors que les deux autres exons le sont. Les exons sont des régions codantes donc importantes. Il est habituel qu’ils soient
remarquablement bien conservés.
La figure 9 présente
un graphique complet du score de conservation en fonction de la position selon
la séquence humaine alignée (contenant des gaps).
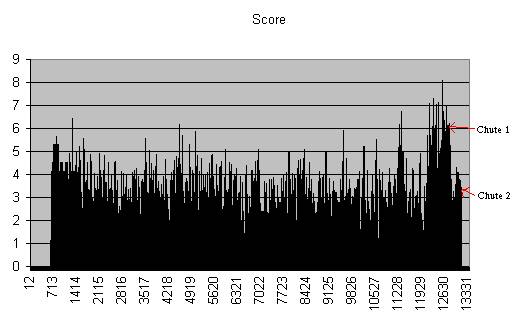
Figure 9. Score de conservation entre la région
englobant le gène humain C12orf18 et ses orthologues chez la souris et le rat
en fonction de la position du nucléotide sur la séquence humaine alignée
(contenant des gaps). (Chute 1.)
Première chute du score conservation dans la région 3’. Voir le texte. (Chute 2.) Deuxième chute du score de conservation dans la région
3’. Voir le texte.
Nous savons que la
région non-traduite 3’ (après l’exon 3) s’étend des positions 12 630 à 12
797. Les figures 8 et 9 illustrent la
conservation de cette région. Selon le
fichier des résultats, on remarque une première chute du score de conservation
dans la région 3’ autour de la position 12 785. Ce qui correspond environ à la fin de la région non-traduite
3’. Nous pouvons en conclure que la
région 3’ non-traduite est bien conservée entre ces espèces.
La poly-adénylation
est l’adjonction d’une extrémité poly-adénylée (poly-A) à l’extrémité 3’ de
l’ARN messager. Elle se produit durant
la maturation de l’ARN messager et seulement chez les eucaryotes (4). La grande région du C12orf18 contient un
signal de poly-adénylation situé environ 300 pb après la fin de l’exon 3
(8). Ce signal est bien conservé entre
les espèces étudiées (8). Cette
position correspond environ à la position 13 131 sur notre séquence alignée
(avec gaps). Selon le fichier des
résultats et la figure 9, on remarque une deuxième chute du score de
conservation dans la région 3’ autour de la position 13 135. Ce qui correspond environ à l’emplacement
approximatif du signal de poly-adénylation.
Nous pouvons en conclure que le signal de poly-adénylation est bien
conservé.
La conservation du
signal de poly-adénylation et de la région non-traduite mais transcrite en 3’
peut indiquer un patron similaire de régulation de l’épissage et de stabilité
de l’ARN messager (8).
7.5
Critique de nos
résultats
Ce que nos résultats
ne permettent pas d’étudier facilement est la conservation des sites de liaison
aux facteurs de transcription. Ces
sites sont trop petits comparativement à la fenêtre coulissante. Pourtant, Dr Blanchette a réussi à en
étudier dans certains cas. Voir la
figure 3. Certains de ces sites sont
conservés dans la régions 5’ proximale du gène C12orf18 selon la littérature
(8).
7.6
Conclusion
Nous pouvons
conclure que l’algorithme fonctionne bien sur notre jeu de données puisqu’il
permet d’identifier comme conservées des régions dont la conservation a été
prouvée par d’autres moyens (8). Notre
expérience montre que l’algorithme fonctionne aussi pour une région beaucoup
plus petite (environ 1 gène) que le CFTR étudié par Dr Blanchette et ses
collègues.
L’algorithme nous a
permis d’identifier des régions conservées dans la grande région du gène
C12orf18: l’exon 2, l’exon 3, la région 3’ non-traduite et la région précédant
le signal de poly-adénylation. De plus,
il nous a donné un indice sur une fonction possiblement conservée de la région
3’ non-codante (la région 3’ non-traduite et la signal de
poly-adénylation). La fonction en
question serait un certain patron (nous ne savons pas lequel) de régulation de
la maturation et de stabilité de l’ARN messager du gène C12orf18 partagé par
l’humain, la souris et le rat.
8
Bibliographie
1. Woese 87.
2. Blanchette, M. Detection and Characterization of Non-Coding Functional Regions of the Human Genome. Conférence.
3. Margulies, E. H., Blanchette, M., NISC Comparative Sequencing Program, Haussler, D. and Green, E. D. 2003. Identification and Characterization of Multi-Species Conserved Sequences. Genome Research,vol. 13, 2003, 2507-2518.)
4. Bernot, A. Analyse de Génomes, Transcriptomes et Protéomes (3e édition). Éd. Dunod (2001).
5. Blanchette, M. and Tompa, M. FootPrinter:
a program designed for phylogenetic footprinting. Nucleic Acids Research, vol. 31, no.
13, 2003, 3840-3842.
6. Blanchette, M. and Tompa, M. Discovery of
Regulatory Elements by a Computational Method for Phylogenetic Footprinting.
Genome Research, vol. 12, no. 5, May
2002, 739-748.
7. Blanchette, M., Schwikowski, B., and Tompa, M. Algorithms
for Phylogenetic Footprinting. Journal of Computational Biology,
vol. 9, no. 2, 2002, 211-223.
- Sargsyan, E., Baryshev, M., Backlund, M., Sharipo, A.,
Mkrtchian, S. Genomic organization
and promoter characterization of the gene encoding a putative endoplasmic
reticulum chaperone, Erp29. Gene, vol. 285, 2002, 127-139.
Remerciements
Mathieu Blanchette.
Alexandre McGrath pour
le fond d’écran.